
Abstract
Antigen recognition by immunoglobulins depends upon initial rearrangements of heavy chain V, D, and J genes. In leporids, a unique system exists for the VH genes usage that exhibit highly divergent lineages: the VHa allotypes, the Lepus sL lineage and the VHn genes. For the European rabbit (Oryctolagus cuniculus), four VHa lineages have been described, the a1, a2, a3 and a4. For hares (Lepus sp.), one VHa lineage was described, the a2L, as well as a more ancient sL lineage. Both genera use the VHn genes in a low frequency of their VDJ rearrangements. To address the hypothesis that the VH specificities could be associated with different environments, we sequenced VDJ genes from a third leporid genus, Sylvilagus. We found a fifth and equally divergent VHa lineage, the a5, and an ancient lineage, the sS, related to the hares’ sL, but failed to obtain VHn genes. These results show that the studied leporids employ different VH lineages in the generation of the antibody repertoire, suggesting that the leporid VH genes are subject to strong selective pressure likely imposed by specific pathogens.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
For the generation of the primary antibody repertoire, most mammals express a varying number of rearranged VDJ genes (Flajnik 2002). The European rabbit (Oryctolagus cuniculus) has a unique system in that, despite having more than 200 VH genes (Ros et al. 2004), it predominantly expresses just one IGHV, the most D-proximal VH1 gene, to generate 80–90 % of its antibody repertoire (Knight and Becker 1990; Knight 1992). The immunoglobulin (Ig) molecules derived from the VH1 express the so-called VHa allotypic markers (e.g. Kindt 1975; Margolies et al. 1977), with three serological lineages having been described in the domestic rabbit, the a1, a2 and a3. These are highly divergent (±20 % amino acid sequence differences), with the allelic specificities of a1 and a2 being correlated with several amino acid differences in framework regions (FR) 1 and 3 (Tonnelle et al. 1983; Mage et al. 1984; Knight and Becker 1990). A fourth equally divergent allotypic lineage, the a4, was described in wild European rabbit Iberian populations (Esteves et al. 2004). The remaining 10–20 % of Ig molecules are encoded by the VHn genes, VHx, VHy and VHz (Kim and Dray 1973; Horng et al. 1976; Roux 1981), that map at least 100 Kb upstream of VH1 (Mage et al. 2006) and do not express VHa allotype-specific determinants.
Studies on the IGHV locus diversity for other leporids are limited to a few data obtained for some Lepus species. Serological analysis conducted for Lepus americanus showed cross-reaction with rabbit anti-a1, anti-a2 and anti-a3 antisera (van der Loo 1987). As for Lepus europaeus and Lepus granatensis, a serological analysis of several populations revealed two phenotypes: partial reaction to anti-a2 antisera and no reaction to any rabbit antiserum (Esteves 2003). Sequencing of rearranged VH genes for Lepus specimens, some of which expressed a2-cross-reacting serum proteins, proved that the a2 polymorphism is trans-specific. The Lepus a2L lineage showed some of the most outstanding characteristics of the rabbit VHa genes such as the presence of five out of 11 amino acid residues that characterize the allotype VHa2 (Mage et al. 1984), and being more closely related to the rabbit VH1-a2 allele than VH1-a2 was to its allelic counterparts VH1-a1 and VH1-a3 (Esteves et al. 2005). A second lineage, the sL, apparently a Lepus-specific lineage, groups distinctively apart from all other VH lineages present in rabbit, showing as distinctive characters the ancestral 70SVK72 motif and residues A10 and K95 (IMGT numbering, Lefranc et al. 2003) (Esteves et al. 2005). More recently, it was shown that, like rabbits, Lepus uses the VHn genes in 5–10 % of its VDJ rearrangements revealing that the antibody repertoire is subject to selective pressure and that the low-frequency usage of VHn genes could be a remnant of an ancient leporid immunologic response to pathogens (Pinheiro et al. 2011, 2013).
Considering that evidence suggests that the leporid VH lineages used to generate the antibody repertoire are subject to selective pressure, in this study, we addressed the hypothesis that the VH lineages represented in the antibody repertoire could be different in species that inhabit different environments due to exposure to different pathogens. Family Leporidae comprises 11 genera, among which Sylvilagus occupies an intermediate position between Oryctolagus and Lepus (Alves and Hackländer 2008), having diverged from Oryctolagus 10 million years ago and from Lepus 12 million years ago (Matthee et al. 2004). Oryctolagus and the two Lepus species, L. europaeus and L. granatensis, for which VDJ genes nucleotide sequences have been obtained, inhabit the Eurasian continent while Sylvilagus is a Native American leporid (Chapman et al. 1980; Flux and Angermann 1990). Starting in 1966, eastern cottontails were introduced in northern Italy through a series of massive releases for hunting purposes; nowadays, eastern cottontails are widespread in this region (Vidus-Rosin et al. 2008). Herein, we sequenced Sylvilagus floridanus VDJ-rearranged genes.
Material and methods
Samples, amplification and sequencing of rearranged VDJ genes
Spleen samples were collected from four eastern cottontail (S. floridanus) specimens collected in Pistoia Province, Italy. Total RNA was extracted using RNeasy Mini Kit (Qiagen, Hilden, Germany), following first-strand complementary (c)DNA synthesis using the iScript cDNA Kit (Bio-Rad, Hercules, CA, USA). VDJ-rearranged genes were PCR amplified using primers VH (5′GGAGACTGGGCTGCGCTGGCTTCTCCTGGT3′; Esteves et al. 2005) and JH2 (5′TGAGGAGACGGTGACCAGGGTGCCT3′; Pinheiro et al. 2013). PCR amplification conditions were as follows: 4 min at 95 °C followed by 35 cycles at 95 °C (45 s), 62 °C (45 s) and 72 °C (60 s), with a final extension at 72 °C (5 min). PCR products were purified (MinElute PCR Purification Kit, Qiagen, Hilden, Germany) and cloned into the pGEM-T Easy vector system II (Promega, Madison, WI, USA).
Phylogenetic analyses
To perform phylogenetic analyses, available sequences for leporid VDJ genes were taken from GenBank. Used sequences include rabbit germ line VH1 and VH4 genes of a1, a2 and a3 allotypes; rabbit cDNA VH gene sequences representative of allotypes a1, a2, a3, a4.1 and a4.2; rabbit germ line and rabbit and Lepus cDNA VH gene sequences of VHx, VHy and VHz and Lepus cDNA VH gene sequences representative of lineages a2L, sLe and sLg. VH genes of the human VH3 family were used as an outgroup. These were aligned with sequences obtained in this study using CLUSTAL W (Thompson et al. 1994) as implemented in BioEdit software (Hall 1999) and the amino acid sequences inferred. Accession numbers for all sequences are given in Table 1.
The computer program jModelTest v2.1.1 (Darriba et al. 2012) was used to assess the fit of our dataset to 88 models of sequence evolution considering the corrected Akaike information criterion (AICc). The phylogenetic relationships were analysed in a Bayesian inference (BI) framework, specifying as optimal mutation model K80+G, retrieved from jModelTest. BI analyses were performed using BEAST v1.7.4 (Drummond and Rambaut 2007). The BEAUti application, part of BEAST package, was used to create the input file for *BEAST. Posterior phylogenies were determined using an uncorrelated lognormal relaxed clock (Drummond et al. 2006) and the Yule tree prior. All parameter priors were set to the defaults. Three independent runs of 50 × 106 generations were performed, sampling trees and parameters every 5 × 103 generations, and concatenated using LogCombiner. Convergence was checked using Tracer v1.5 (Rambaut and Drummond 2007), and summary trees were generated with TreeAnnotator v1.5.4, part of the BEAST package.
Genetic distances between VH genes among species were calculated using the “compute net average distance between groups” option of MEGA5.0 software (Tamura et al. 2011). This option corrects for variance due to differences within groups, as expected in VH-rearranged genes which are somatically diversified.
Results
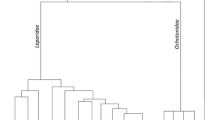
A total of 86 VDJ gene sequences were obtained for the four studied S. floridanus individuals. Phylogenies were estimated with the whole VH sequence and excluding the CDRs, yielding similar clustering of the analysed sequences. Thus, we present here the phylogeny obtained with the whole VH sequence (Fig. 1). Clusters of the previously described rabbit and hare lineages are shown with good support on the phylogenetic tree: the VHn lineage (1.00 posterior probability), the sL lineage (1.00 posterior probability) and the VHa cluster (0.96 posterior probability) that includes the VHa1, VHa2, VHa2L, VHa3 and VH4 lineages (1.00, 1.00, 0.99, 1.00 and 1.00 posterior probabilities, respectively). The S. floridanus sequences obtained in this study fall into two groups that include only Sylvilagus VH sequences: (i) a cluster that is not related to any previously described leporid lineages, the sS lineage (1.00 posterior probability), and (ii) a second group that clusters with the rabbit VHa lineages, designated here as a5 lineage (1.00 posterior probability). This a5 lineage is further subdivided into two groups, the a5.1 and a5.2 variants (0.98 and 0.95 posterior probabilities, respectively).

Phylogenetic tree of leporid VH genes. Human VH clan 3 genes were used to root the tree. Groups were collapsed for simplification; the number of sequences in each group is indicated. The a1, a2, a3 and a4 groups include only European rabbit sequences, the a2L and sL groups include only hare sequences and the a5.1, a5.2 and sS groups include only S. floridanus sequences. The VHn group includes European rabbit and hares sequences. BI posterior probabilities are depicted in front of each node
Of the 86 obtained sequences, 46 were classified as a5.1, 26 were classified as a5.2 and 14 sequences were classified as sS. These three groups appear to have an allelic distribution as each one of the analysed individuals has sequences belonging to two of the groups: the Sfl1 and Sfl4 specimens have sequences that were classified as a5.1 and sS (13–9 and 11–5) and the Sfl2 and Sfl3 specimens have sequences that were classified as a5.1 and a5.2 (2–17 and 17–9).
Genetic distances between rabbit, hares and cottontail VH genes
The genetic distances between S. floridanus sS lineage and the a1–a4 lineages (0.12–0.16, Table 2) are identical to those between the hares sL lineage and the VHa1–VHa4 lineages (0.10–0.16, Table 2) or between the VHn and the VHa1–VHa4 lineages (0.08–0.15, Table 2). The S. floridanus a5 lineage genetic distances to other VHa lineages (0.06–0.10, Table 2) are identical to the genetic distances observed between VHa1 and VHa4 lineages (0.07–0.14, Table 2). The genetic distance between the a5.1 and a5.2 variants is lower than the genetic distance between a4.1 and a4.2 variants (0.03 and 0.06, respectively).
Characterization of expressed VH in S. floridanus
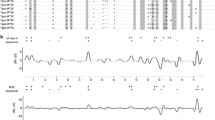
The inferred amino acid sequences were grouped according to the phylogenetic clustering and compared to Lepus cDNA and rabbit germ line and cDNA VH genes representative of VHn and VHa allotypes as well as sequences representative of lineages a2L and sL (Fig. 2).

Alignment of leporid VH protein sequences. a European rabbit and hares VHa lineages and S. floridanus a5.1 lineage. b S. floridanus a5.2 and sS lineages, hares sL lineage and European rabbit and hares VHn lineage. Dashes represent alignment gaps. Dots indicate identity with the master sequence except at gap positions. The “IMGT Protein display for V domain” header is shown (Lefranc et al. 2003). Sequences are grouped into phylogenetic groups according to the tree in Fig. 1. Shown are representatives of European rabbit VH1a1, VH1a2, VH1a3, Vhx, Vhy and Vhz germ line sequences (GenBank accession numbers M93171, M93172, M93173, L03846, L03890 and AF264469, respectively); European rabbit a4.1 and a4.2 cDNA sequences (GenBank accession numbers AY207980 and AY207967) and hares a2L and sL cDNA sequences (GenBank accession numbers AY288450, AY288465 and AY288459). Also shown are all S. floridanus cDNA sequences obtained in this study that were grouped into the a5.1, a5.2 and sS lineages (for GenBank accession numbers please see Table 1). Highlighted in grey are the VHa deletion at position 2 and motifs 19LTLTCT24 and 70WAK72, and sL/sS sequences 70SVK72 motif. Also highlighted are residue characteristics of a5 and sS lineages
The rabbit VHa sequences are characterized by the 19LTLTCT24 of FR1 and 70WAK72 of FR3 motifs as well as showing a deletion at position 2 (Mage et al. 1984). The 72 sequences obtained in this study that were classified as VHa have the deletion at position 2, and the majority either has the characteristic 70WAK72 or a 70WAQ72 motif. The 19LTLTCT24 of FR1 is also present though the majority of the sequences have a D20 substitution. These sequences further have as characteristics the motif 83SAG85 and residue D96, while also sharing the VHa2 I/F13 and T79 residues. Thus, these sequences have the rabbit VHa hallmark residues but have evolved some particular features which substantiate their inclusion in a new VHa lineage that we designate as a5. According to the phylogenetic tree, this a5 lineage has two variants, the a5.1 and a5.2, and amino acid differences exist between them that support their distinction. The 19LTLTCT24 motif is more conserved in the sequences assigned as a5.2 while those that were classified as a5.1 have the D20 as well as K24 substitutions. In FR3, the 83SAG85 D96 motif is more conserved in the sequences that were classified as a5.1 while those identified as a5.2 have K80, L87, G93 and E97 residues that, although not a characteristic to this variant, are not present in the a5.1 variant.
The 14 sequences that were classified as a new lineage (sS) have the ancestral 70SVK72, also present in the hares sL lineage, and the 2EQ3 motif of rabbit VHx peptides. The FR3 amino acid sequence between residues 83 and 101 has nine substitutions that are characteristics to this lineage: 83DA84, 87LE88, D93, 95KTT97 and I101.
Discussion
The generation of antibody repertoire in the European rabbit has been widely studied, and several particularities have been found. It seems to rely on a reduced number of VH genes: the VH1, which expresses the so-called VHa allotypic markers, and the VHx, VHy and VHz genes, or VHn genes, which do not have the VHa allotype specificities (Mage et al. 1984; Knight and Becker 1990). For the European rabbit, four highly divergent allelic lineages have been described for the VHa allotypes: a1, a2, a3 and a4 (Mage et al. 1984; Knight and Becker 1990; Esteves et al. 2004). In hares (Lepus sp.), the existence of a VHa lineage, a2L, with nucleotide and amino acid sequence similarity with the rabbit a2 lineage and which shows cross-reaction with sera against the rabbit a2 lineage has showed the trans-specific nature of the VHa2 polymorphism (Esteves et al. 2005). A more ancestral lineage, sL, presumably also derived from the VH1 gene, was found to be expressed in hares (Esteves et al. 2005). More recently, Pinheiro and co-workers (2013) showed that hares and rabbits share the low-frequency usage of the VHn genes in the generation of the antibody repertoire.
In this study, we observed VH-expressed genes for S. floridanus that have the rabbit VHa signature, lineage a5, as well as VH-expressed genes that belong to a more ancestral lineage, sS, that seems to be S. floridanus specific. In the phylogenetic tree, the S. floridanus a5 sequences group with rabbit and hare VHa lineages, clearly apart from the S. floridanus sS sequences, and the genetic distances also indicate that the a5 lineage is more similar to the VHa lineages than to the sS lineage. These results suggest that the VHa allotypes have an origin that pre-dates the leporid radiation. Some VHa lineages originated before the species split and are shared between species, such as the a2 and a2L lineages, whereas others apparently are species specific. As Esteves et al. (2004) argued, it is likely that evolutionary modes, and mutational rates, vary among VHa lineages having increased in the a1, a2 and a4 lineages, which would explain the large inter-lineage distances despite a more recent origin. An interesting aspect resides in the differences in number of VHa lineages found in each leporid species studied so far. Apparently, the rabbit has retained a greater diversity of VHa lineages, with at least four lineages (Mage et al. 1984; Knight and Becker 1990; Esteves et al. 2004), while hares and cottontail have just one lineage each (Esteves et al. 2005; Pinheiro et al. 2013; present study). This higher number of VHa lineages found in rabbit can be the result of a compensatory mechanism due to the loss of a more ancient lineage of VH genes, which would be the rabbit equivalent of the hares sL and cottontail sS. These ancient lineages, characterized by the motif 70SVK72 and more highly divergent from the VHa lineages, can importantly contribute to the diversity of the antibody repertoire. Its loss may have imposed the diversification of the VHa lineages in rabbit. Contrarily to what is widely accepted, increasing evidence suggests the VH lineages which are rearranged in different leporid species are under different selective pressures, either driven by pathogens or by the self, though it is not evident how this selection operates (Pinheiro et al. 2013). It has also been shown that in rabbit, the diversification of the antibody repertoire requires the interaction with exogenous factors such as the gut microbiota (Lanning et al. 2000a, b; Sehgal et al. 2002), which may also exert selective pressure on the expressed VH lineages.
Sequencing of random germ line VH genes has shown that VHn genes are present in S. floridanus genome (Esteves 2003); thus, we hypothesized that, similarly to what was found for rabbit and hares (Pinheiro et al. 2013), S. floridanus would use these genes in a low percentage of VDJ rearrangements. Interestingly, despite the identical sequencing effort made for Lepus and Sylvilagus rearranged VH (circa 25 sequences per individual; Pinheiro et al. 2013; present study), we failed to recover rearranged VDJ genes with characteristics of rabbit and hares VHn genes in the studied S. floridanus specimens. S. floridanus may not use the VHn genes in the generation of the antibody repertoire or may do so in such a low percentage (under 4 %) that the sequencing effort would have to be greatly increased to allow its detection. The usage of VHn genes in low frequency both in rabbit (Kim and Dray 1973; Roux 1981) and hares shows that, in leporids, the VH lineages used to generate the antibody repertoire are subject to selective pressure (Pinheiro et al. 2013). The rabbit and hare species for which the expression of VHn has been shown, L. europaeus, are European leporids while S. floridanus is an American leporid (Chapman et al. 1980; Flux and Angermann 1990). Interestingly, two related caliciviruses affect the European rabbit and two hare species of Eurasian distribution (L. europaeus and Lepus timidus) but not cottontails (Abrantes et al. 2012; Lopes et al. 2013). Thus, we can put forward the hypothesis that the different continents’ pathogen community may impose different selective pressures and favour the expression of VHn only in European species.
References
Abrantes J, van der Loo W, Le Pendu J, Esteves PJ (2012) Rabbit haemorrhagic disease (RHD) and rabbit haemorrhagic disease virus (RHDV): a review. Vet Res 43(1):12
Alves PC, Hackländer K (2008) Lagomorph species: geographical distribution and conservation status. In: Alves PC, Ferrand N, Hackländer K (eds) Lagomorph biology. Springer, Berlin, pp 395–405
Chapman JA, Hockman JG, Ojeda MM (1980) Sylvilagus floridanus. Mamm Species 136:1–8
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9(8):772
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Drummond AJ, Ho SY, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4(5):e88
Esteves PJ (2003) Molecular and population genetic analysis of polymorphism at the antibody loci IgGCH2 and IgVH in lagomorphs. Faculdade de Ciências Universidade do Porto, Porto (http://hdl.handle.net/10216/10598)
Esteves PJ, Lanning D, Ferrand N, Knight KL, Zhai SK, van der Loo W (2004) Allelic variation at the VHa locus in natural populations of rabbit (Oryctolagus cuniculus, L.). J Immunol 172(2):1044–1053
Esteves PJ, Lanning D, Ferrand N, Knight KL, Zhai SK, van der Loo W (2005) The evolution of the immunoglobulin heavy chain variable region (IgVH) in Leporids: an unusual case of transspecies polymorphism. Immunogenetics 57(11):874–882
Flajnik MF (2002) Comparative analyses of immunoglobulin genes: surprises and portents. Nat Rev Immunol 2(9):688–698
Flux JEC, Angermann R (1990) The hares and jackrabbits. In: Chapman JA, Flux JEC (eds) Rabbits, hares and pikas: status survey and conservation action plan. The World Conservation Union, Gland, pp 61–94
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Horng WJ, Knight KL, Dray S (1976) Heavy chain variable region allotypic sub-specificities of rabbit immunoglobulins. I. Identification of three subpopulations of a1 IgG molecules. J Immunol 116(1):117–125
Kim BS, Dray S (1973) Expression of the a, x, and y variable region genes of heavy chains among IgG, IgM, and IgA molecules of normal and a locus allotype-suppressed rabbits. J Immunol 111(3):750–760
Kindt TJ (1975) Rabbit immunoglobulin allotypes: structure, immunology, and genetics. Adv Immunol 21:35–86
Knight KL (1992) Restricted VH gene usage and generation of antibody diversity in rabbit. Annu Rev Immunol 10:593–616
Knight KL, Becker RS (1990) Molecular basis of the allelic inheritance of rabbit immunoglobulin VH allotypes: implications for the generation of antibody diversity. Cell 60(6):963–970
Lanning D, Sethupathi P, Rhee KJ, Zhai SK, Knight KL (2000a) Intestinal microflora and diversification of the rabbit antibody repertoire. J Immunol 165(4):2012–2019
Lanning D, Zhu X, Zhai SK, Knight KL (2000b) Development of the antibody repertoire in rabbit: gut-associated lymphoid tissue, microbes, and selection. Immunol Rev 175:214–228
Lefranc MP, Pommié C, Ruiz M, Giudicelli V, Foulquier E, Truong L, Thouvenin-Contet V, Lefranc G (2003) IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol 27(1):55–77
Lopes AM, Gavier-Widén D, Le Gall-Reculé G, Esteves PJ, Abrantes J (2013) Complete coding sequences of European brown hare syndrome virus (EBHSV) strains isolated in 1982 in Sweden. Arch Virol 158(10):2193–6
Mage RG, Bernstein KE, McCartney-Francis N, Alexander CB, Young-Cooper GO, Padlan EA, Cohen GH (1984) The structural and genetic basis for expression of normal and latent VHa allotypes of the rabbit. Mol Immunol 21(11):1067–1081
Mage RG, Lanning D, Knight KL (2006) B cell and antibody repertoire development in rabbits: the requirement of gut-associated lymphoid tissues. Dev Comp Immunol 30(1–2):137–153
Margolies MN, Cannon LE, Kindt TJ, Fraser B (1977) The structural basis of rabbit VH allotypes: serologic studies on a1 H chains with defined amino acid sequence. J Immunol 119(1):287–294
Matthee CA, van Vuuren BJ, Bell D, Robinson TJ (2004) A molecular supermatrix of the rabbits and hares (Leporidae) allows for the identification of five intercontinental exchanges during the Miocene. Syst Biol 53(3):433–447
Pinheiro A, Lanning D, Alves PC, Mage RG, Knight KL, van der Loo W, Esteves PJ (2011) Molecular bases of genetic diversity and evolution of the immunoglobulin heavy chain variable region (IGHV) gene locus in leporids. Immunogenetics 63(7):397–408
Pinheiro A, de Mera IG, Alves PC, Gortázar C, de la Fuente J, Esteves PJ (2013) Sequencing of modern Lepus VDJ genes shows that the usage of VHn genes has been retained in both Oryctolagus and Lepus that diverged 12 million years ago. Immunogenetics 65(11):777–84
Rambaut A, Drummond AJ (2007) Tracer v1.5. Available from http://beast.bio.ed.ac.uk/Tracer
Ros F, Puels J, Reichenberger N, van Schooten W, Buelow R, Platzer J (2004) Sequence analysis of 0.5 Mb of the rabbit germline immunoglobulin heavy chain locus. Gene 330:49–59
Roux KH (1981) A fourth heavy chain variable region subgroup, w, with 2 variants defined by an induced auto-antiserum in the rabbit. J Immunol 127(2):626–632
Sehgal D, Obiakor H, Mage RG (2002) Distinct clonal Ig diversification patterns in young appendix compared to antigen-specific splenic clones. J Immunol 168(11):5424–5433
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tonnelle C, Cazenave PA, Brezin C, Moinier D, Fougereau M (1983) Structural correlates to the rabbit immunoglobulin heavy chain a100 allotype. Mol Immunol 20(7):753–761
van der Loo W (1987) Population genetical studies on the adaptive significance of the immunoglobulin allotypes in wild rabbit. In: Dubiski S (eds) The rabbit in contemporary immunological research. Longman Scientific & Technical, John Wiley, pp 101–126
Vidus Rosin A, Gilio N, Meriggi A (2008) Introduced Lagomorphs as a threat to “native” Lagomorphs: the case of the Eastern cottontail (Sylvilagus floridanus) in northern Italy. In: Alves PC, Ferrand N, Hacklander K (eds) Lagomorph biology. Evolution, ecology and conservation. Springer, Eidenberg, pp 153–165
Acknowledgments
This work was partially funded by FEDER (Fundo Europeu de Desenvolvimento Regional) funds through the Programa Operacional Factores de Competitividade (COMPETE program; FCOMP-01-0124-FEDER-028286) and Portuguese national funds through FCT (Fundação para a Ciência e a Tecnologia; research project PTDC/BIO-ANM/3963/2012)—Quadro de Referência Estratégico Nacional (QREN) funds from the European Social Fund and Portuguese Ministério da Educação e Ciência. FCT also supported the doctoral grant of AP (ref.: SFRH/BD/71252/2010) and the FCT Investigator grant of JA (ref.: IF/01396/2013) and the Post-Doctoral grant of JMF (ref.: SFRH/BPD/43264/2008). “Genomics Applied to Genetic Resources” co-financed by North Portugal Regional Operational Programme 2007/2013 (ON.2 – O Novo Norte), under the National Strategic Reference Framework (NSRF), through the European Regional Development Fund (ERDF), also supported this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pinheiro, A., Melo-Ferreira, J., Abrantes, J. et al. Sequencing of Sylvilagus VDJ genes reveals a new VHa allelic lineage and shows that ancient VH lineages were retained differently in leporids. Immunogenetics 66, 719–726 (2014). https://doi.org/10.1007/s00251-014-0807-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-014-0807-0