
Abstract
Multiple factors determine the ability of a peptide to elicit a cytotoxic T cell lymphocyte response. Binding to a major histocompatibility complex class I (MHC-I) molecule is one of the most essential factors, as no peptide can become a T cell epitope unless presented on the cell surface in complex with an MHC-I molecule. As such, peptide-MHC (pMHC) binding affinity predictors are currently the premier methods for T cell epitope prediction, and these prediction methods have been shown to have high predictive performances in multiple studies. However, not all MHC-I binders are T cell epitopes, and multiple studies have investigated what additional factors are important for determining the immunogenicity of a peptide. A recent study suggested that pMHC stability plays an important role in determining if a peptide can become a T cell epitope. Likewise, a T cell propensity model has been proposed for identifying MHC binding peptides with amino acid compositions favoring T cell receptor interactions. In this study, we investigate if improved accuracy for T cell epitope discovery can be achieved by integrating predictions for pMHC binding affinity, pMHC stability, and T cell propensity. We show that a weighted sum approach allows pMHC stability and T cell propensity predictions to enrich pMHC binding affinity predictions. The integrated model leads to a consistent and significant increase in predictive performance and we demonstrate how this can be utilized to decrease the experimental workload of epitope screens. The final method, NetTepi, is publically available at www.cbs.dtu.dk/services/NetTepi.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cytotoxic T lymphocytes (CTL) play a critical role in cell-mediated immunity by scanning peptides presented by major histocompatibility complex class I (MHC-I) molecules on the cell surface. The peptides bound by MHC-I molecules are prevalently 8–11 residues in length and derived from intracellular proteins. During protein degradation, peptides are sampled by MHC-I molecules and transported to the cell surface. CTL recognition of a peptide-MHC (pMHC) with a bound non-self peptide triggers the release of cytotoxic effector proteins, inducing the infected cell to undergo apoptosis (Harty et al. 2000).
Knowledge of which peptides within an intracellular pathogen are most likely to elicit an immune response is essential for the development of reagents, therapeutics, and diagnostic tools. These immunogenic peptides, referred to as T cell epitopes, can be identified experimentally, but due to the large number of unique peptides in a virus or bacterial proteome, epitope screens commonly incorporate a prefiltering step using an in-silico T cell epitope prediction method (reviewed in Lundegaard et al. 2012). Due to pMHC binding being the most selective factor for potential T cell epitopes, this aspect has been studied extensively (Yewdell and Bennink 1999). More than 150,000 distinct pMHC binding affinity measurements are contained in the Immune Epitope Database (IEDB, Vita et al. 2010) facilitating the development of high performance pMHC binding affinity prediction methods. The best performing of these include NetMHC (Nielsen et al. 2003; Lundegaard et al. 2008a), NetMHCpan (Nielsen et al. 2007; Hoof et al. 2009), NetMHCcons (Karosiene et al. 2012), and SMM (Peters and Sette 2005), all hosted at the IEDB Analysis Resource (Kim et al. 2012).
While all T cell epitopes must be MHC binders, not all MHC binders are T cell epitopes (Feltkamp et al. 1994). This observation has prompted several studies to investigate what additional factors are required for a peptide to be immunogenic. Earlier studies have analyzed the impact of including predictions of other MHC-I pathway players, such as proteasomal cleavage (Keşmir et al. 2002; Nielsen et al. 2005) and TAP transport (Peters et al. 2003), on the ability to correctly identify CTL epitopes (Tenzer et al. 2005; Larsen et al. 2005; Larsen et al. 2007; Stranzl et al. 2010). While results from these analyses are not fully consistent in terms of the relative importance of the different players, they all concur in the finding that additional features beyond antigen processing and MHC binding affinity are determinants of peptide immunogenicity.
Recent studies have investigated the role of pMHC stability in determining whether a peptide is a T cell epitope (Harndahl et al. 2012; Jørgensen et al. 2014). Whereas pMHC binding affinity defines the equilibrium between the association and dissociation of the pMHC complex, pMHC stability defines the rate of dissociation. The more stable the pMHC, the longer a peptide is bound to the MHC molecule once loaded. The rationale behind the importance of pMHC stability is that the complex must stay intact during transportation to the cell surface, and remain so on the cell surface, while waiting for a naïve CD8+ T cell to bind it. The number of naïve CD8+ T cells in a host specific for a given pMHC is extremely low, so the longer a pMHC is stable on the surface of the cell, the higher the chance of being recognized by a T cell and initiating an immune response. Following this argument, Jørgensen et al. (2014) showed that a combination of binding affinity and stability predictions had a greater predictive performance for T cell epitope identification than either prediction type alone. One should note that on-rates could be considered as important as dissociation in determining peptide immunogenicity, as they dictate which peptides will end up on the surface of the presenting cell. However, to the best of our knowledge no high throughput experimental and/or prediction methods are currently available to estimate peptide on-rates in situations corresponding to a live cell, making assessment of this aspect of antigen presentation difficult.
In addition to being presented on an MHC molecule, it is also required that the peptide is recognized by a T cell receptor (TCR) in order to elicit an immune response. As such, TCR binding has been studied extensively (reviewed by Rudolph et al. 2006). For pMHC binding, it has been shown that some positions in the peptide, termed anchor positions, have a larger impact on binding affinity than others, and that different MHC molecules have different amino acid residue preferences at these positions (Falk et al. 1991). Similarly, certain positions in the peptide have a larger impact on TCR binding compared to others (Lee et al. 2004; Frankild et al. 2008). In general, positions 4–6 are found to have the highest importance as mutations in these positions affect TCR binding the most, but other positions may also have an impact if they affect the peptide conformation in the MHC binding pocket (Tynan et al. 2005). The role of T cell cross-reactivity on peptide immunogenicity has also been investigated, suggesting that TCRs recognize peptides with similar biochemical properties (Frankild et al. 2008). This has implications for peptide immunogenicity, as TCRs that recognize self-pMHCs are negatively selected during T cell maturation (Huseby et al. 2005), leaving “holes” in the T cell repertoire. Predicting pMHC binders that are biochemically similar to self peptides has proven to be a viable method for distinguishing non-immunogenic peptides from T cell epitopes (Frankild et al. 2008; Calis et al. 2012).
Tung et al. have developed a method for the prediction of peptide immunogenicity, POPISK (Tung et al. 2011). The method uses support vector machines to predict T cell reactivity and identify important positions for TCR binding in a data driven manner. The method was trained only on HLA-A02:01-restricted epitopes, which means predictions are only available for that allele. Similarly, Calis et al. (2013) have recently developed an immunogenicity model for the prediction of a peptide’s T cell propensity. An important difference between the two methods is that the latter was trained on epitopes from 12 MHC-I molecules (six mouse and six human) with MHC anchor positions disregarded, meaning the model can provide predictions for peptides presented by any MHC-I molecule. The model suggests that positions 4–6 in the peptide are the most important for TCR binding, consistent with previous studies (Frankild et al. 2008). The model also suggests that the TCR has a preference for residues with large or aromatic side chains, consistent with previous studies that have shown an association between immunogenicity and the presence of large, aromatic amino acid residues (Alexander et al. 1994).
In this study, we extend the work by Jørgensen et al. (2014) with the aim of creating an integrated method for T cell epitope prediction, combining pMHC binding affinity, pMHC stability, and T cell propensity predictions. Using a large set of T cell epitope data obtained from the IEDB and SYFPEITHI (Rammensee et al. 1999), we investigate if the integrated method can outperform each individual prediction method, and to what degree such an improvement in predictive performance can be translated to a reduction in the experimental workload for epitope screens that utilize in-silico prefiltering.
Methods and materials
Model training data
A T cell epitope data set previously compiled by Jørgensen et al. (2014) was used to optimize the weight on each prediction type used in the three combination models. The data set contained 295 T cell epitopes downloaded from SYFPEITHI (Rammensee et al. 1999) and 1,216 T cell epitopes downloaded from the IEDB (Vita et al. 2010), covering nine HLA alleles. All epitopes in this data set were 9mers and had been prefiltered to remove all epitopes that were not predicted to be binders to their annotated HLA molecule by NetMHCcons. Throughout this study, we define an MHC binder as a peptide with either a predicted binding affinity stronger than 500 nM, or a predicted rank score within the top 2 %. The data was further filtered to remove any peptides present in the data used by Calis et al. (2013) to create the T cell propensity model. This removed 310 epitopes (21 %) from the data leaving a total epitope count of 1,201.
An overview of the data set can be found in Table 1. Allele-balanced training data sets were created by randomly selecting 50 epitopes from each of the nine HLA alleles. If less than 50 epitopes were available for an allele, all epitopes were selected. The training data sets contained a total of 378 epitopes.
Evaluation data
T cell epitopes were downloaded from the IEDB to create an evaluation data set. Epitopes were selected based on the following three criteria: (a) 9-10 amino acids in length, (b) annotated to elicit a positive IFN-g response, and (c) annotated to bind to one of the 13 HLA molecules for which stability predictions are currently available in the NetMHCstab method (as of November 2013). 1,428 epitopes were identified and downloaded.
As in the Jørgensen data set, predicted non-binding epitopes were filtered out of the evaluation data set. Affinity predictions for all the epitopes were calculated using NetMHCcons and all epitopes not predicted to be MHC binders were discarded (Jørgensen et al. 2014). This filtered out 273 epitopes (19 %) from the data set. The data was then filtered further to remove any peptides also present in the Calis data set, removing 215 epitopes (15 %). Finally, the evaluation data set was filtered to remove any peptides also present in the model training data set. This removed 375 epitopes (26 %) from the data. Due to the low number of epitopes available for HLA-B27:05 and HLA-B39:01 in the final evaluation data set, these alleles were excluded from the evaluation. The final data set consisted of 557 epitopes covering 11 HLA molecules.
Source proteins for each of the epitopes were downloaded from GenBank using the accession number annotated in the IEDB. An overview of the 557 epitopes can be found in Table 2. All training and evaluation data is available at www.cbs.dtu.dk/suppl/immunology/NetTepi-1.0.
Evaluation methods
Area under the receiver operating characteristic curve (AUC) scores were used to evaluate the performance of the different models. More specifically, AUC0.1 scores, corresponding to a specificity threshold of 0.9 were used, as the highest scoring peptides are usually those of interest for users of epitope prediction methods. Receiver operating characteristic (ROC) curves were calculated by splitting the source protein into overlapping 9 and 10mers where the epitope was considered the sole positive and all other peptides were considered negatives. In cases where a protein contained several known epitopes from the evaluation data set, each epitope-HLA pair formed an individual entry. When evaluating the performance in such cases, the set of negative peptides were filtered for these additional epitopes, removing what would have been a large amount of false positives in the ROC analysis. One-tailed binomial tests (excluding ties), based on the number of instances where a method had higher predictive performance than another, were used to evaluate whether there were significant differences between the performances of the different models.
Model training
The models were fitted on the allele-balanced training data using a 5-fold cross-validation (CV) strategy in which 1/5 of the data is left out as test data and the model is trained on the remaining 4/5. This was repeated three times, each time with a new training data set obtained by randomly selecting up to 50 epitopes for each allele from the training data set. In each run and CV, optimal model parameters were selected based on the highest test performance evaluated using AUC0.1. As the training data consisted of solely 9mer epitopes, only overlapping 9mers were here used as negatives in the ROC analysis. The CV setup thus produced 15 optimal weights for each model and a majority vote was used to select the final weights to be used for further evaluation.
10mers
While NetMHCcons and NetMHCstab are able to make predictions on 8–14mers, T cell propensity predictions using the model described by Calis et al. (2013) were only described for 9mers. We extended the immunogenicity model to support 10mer predictions by using the approximation method proposed by Lundegaard et al. (2008b). In short, the approximation consists of generating six 9mer peptides for each 10mer peptide removing in turn one amino acid at P4, P5, P6, P7, P8, and P9, and next predicting the score of the 10mer peptide as the average score of the six 9mers.
Results
Three prediction types were used to create the integrated T cell epitope prediction method: pMHC binding affinity, pMHC stability, and T cell propensity. pMHC binding affinity predictions were obtained using NetMHCcons-1.0 (Karosiene et al. 2012), a consensus method utilizing optimized combinations of predictions from the NetMHC (Nielsen et al. 2003; Lundegaard et al. 2008a), NetMHCpan (Nielsen et al. 2007; Hoof et al. 2009), and PickPocket (Zhang et al. 2009) methods. pMHC stability predictions were obtained using NetMHCstab-1.0 (Jørgensen et al. 2014), an artificial neural network-based method capable of predicting the half-lives of pMHCs. T cell propensity predictions were calculated using the immunogenicity model proposed by Calis et al. (2013).
Using these prediction types, we constructed three different models, one combining pMHC binding affinity and T cell propensity (AT), another combining pMHC binding affinity and pMHC stability (AS), and finally a model combining all three predictions (AST). In each model, the predictions were combined using a weighted sum:
where t, s, and t + s fall in the range 0–1 and where the NetMHCcons and NetMHCstab scores are the raw prediction values falling in the range 0–1 The model combining pMHC stability with T cell propensity was not included in the study, as this model displayed inferior predictive performance compared to the binding affinity-based model alone (data not shown).
Weight optimization
From the balanced training data set, optimal relative weights for each model were obtained. For the AT model the optimal weight was s = 0.11 +/− 0.037, for the AS model t = 0.15 +/− 0.034, and for the AST model the optimal weights were s = 0.1 +/− 0.035 and t = 0.16 +/− 0.012, were values after the +/− sign are the standard deviations of the 15 optimal weights found in the CV procedure.
Model evaluation
Each model was evaluated on the evaluation data set. An overview of the performances can be found in Table 3. The reported AUC0.1 scores are the averages of the AUC0.1 scores calculated for each epitope-protein pair in the evaluation set. In addition to the three models, performance measures for the individual prediction types are included for reference. Performance measures for the different models on the evaluation data when split into 9 and 10mers, as well as when split into the 11 HLA molecules are also included in Table 3.
Binomial tests were used to test for significant differences in performance. When evaluating on all the data, the AST model was the highest performing model, performing significantly better than all three individual methods (p < 0.001 for each method) and the AT model (p = 0.002), but not the AS model (p = 0.062). The AT and AS models both performed significantly better than NetMHCcons (p = 0.025 and p < 0.001, respectively) as well as NetMHCstab and the immunogenicity model alone (p < 0.001 in all cases).
Looking at the length specific data, the AS model was numerically the highest performing method for 9mers, followed by NetMHCcons, the AST model, and finally the AT model. In terms of the statistical test, on this data set all three models performed significantly better than NetMHCstab and T cell propensity, but only the AS model performed significantly better than NetMHCcons (p = 0.040). For 10mers, the AST model was significantly better than the AT model (p = 0.006) and AS model (p = 0.027), as well as each of the three individual methods (p < 0.001 for each method).
As can be seen in Table 3, the performance gain obtained by adding T cell propensity and pMHC stability predictions to pMHC binding affinity varies from allele to allele, with T cell propensity adding more on some alleles, and pMHC stability adding more on others. The AST model was not numerically the highest performing model for all alleles. It achieved higher performance than NetMHCcons for eight alleles and a higher performance than the AT and AS models for six alleles in each case.
To further quantify the performance difference between the four top performing methods, false positive (FP) ratios for each epitope-protein pair were compared. We define FPs as all peptides in a protein with a higher prediction score than the known epitope, and the FP ratio as the number of FPs normalized by the total number of peptides in the protein. Both 9 and 10mers were included in this analysis.
Figure 1 shows the fraction of epitopes in our evaluation data that would be identified given a certain FP ratio. The insert shows the complete graph, and it can be seen that all four methods achieve highly comparable performance values reaching a sensitivity of 100 % at an FP ratio of around 7 %. The outer plot shows the sensitivity curves up to an FP ratio of 2 %. In the majority of this plot, the yellow line (AST) is on the top demonstrating the improved performance of the AST model. For example, allowing for an FP ratio of 1.5 %, NetMHCcons identifies 88.0 % of the epitopes, whereas the AST model identifies 89.9 % of the epitopes, a 2.1 % increase. Likewise, at a sensitivity of 90 %, NetMHCcons has an FP ratio of 1.7 %, whereas the AST model has an FP ratio of 1.5 %.

Plot showing the sensitivity of each prediction method as a function of the FP ratio. The insert shows the complete graph and the outer plot focuses on the FP ratios from 0 to 0.02
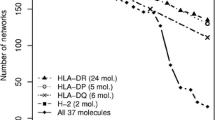
As shown above, the absolute difference in predictive performance between the different methods is relatively small when measured in terms of difference in averaged AUC0.1 and FP ratio values. However, as demonstrated by the significant p values when comparing the combined AST model to the other models, the increase in predictive performance of the AST model is consistent over the majority of the epitopes included in the benchmark analysis. This consistency in increased prediction accuracy can be appreciated by calculating the accumulative number of times each method achieves the highest predictive performance as a function of decreased predicted binding strength. The results of such an analysis are shown in Fig. 2. Here, the epitope-protein pairs were sorted based on the predicted percentile rank binding affinity of the epitopes, and number of top performances (“wins”) for each method was accumulated. All ties were disregarded in this analysis. A similar figure showing the accumulative number of top performances (“wins”) for each method as a function of decreased predicted binding affinity in nM units is shown in Supplementary Figure S1.

Bar-plot showing the accumulated number of wins for each prediction method as a function of the percentile rank score for each epitope in the evaluation data set
The figure clearly demonstrates that in the top rank percentile ranges (strongest binders for each allele), integration of binding stability predictions contribute the most to the improved performance, with the AS model being the top performing method in the 0.01–0.2 rank percentile range. However, due to the very small number of observations in this part of the figure, none of the observed differences are statistically significant. As the rank percentile score is decreased, T cell propensity predictions gain importance and the AST model pulls ahead and achieves most wins, followed by the AT and AS models. Comparing the AST model to NetMHCcons, the AST model has 37 % more wins across the entire evaluation set.
Discussion
No peptide can become a T cell epitope without first being bound and presented by an MHC-I molecule. However, not all peptide binders have the ability to activate CTLs, suggesting that factors other than pMHC binding affinity play a role in defining peptide immunogenicity. Due to pMHC binding being the strongest selective step in CTL activation, pMHC binding affinity prediction methods are currently the go-to tools for T cell epitope prediction (Lundegaard et al. 2010). Still, recent studies have demonstrated that other prediction types are also relevant predictors of T cell epitopes (Frankild et al. 2008; Tung et al. 2011; Calis et al. 2012; Harndahl et al. 2012; Calis et al. 2013; Jørgensen et al. 2014).
In this study, we combined pMHC binding affinity predictions from NetMHCcons, pMHC stability predictions from NetMHCstab, and T cell propensity predictions from the immunogenicity model by Calis et al. (2013) to create three linear integrative models for T cell epitope prediction. One model combines binding affinity and stability, another combines binding affinity and T cell propensity, and the last combines all three prediction types. The relative weights on the prediction types were estimated using an allele-balanced training data set of 9mer T cell epitopes downloaded from the IEDB and SYFPEITHI.
The models and prediction methods were all evaluated on an evaluation data set of 9 and 10mer T cell epitopes downloaded from the IEDB. The immunogenicity model was extended to allow for 10mer predictions using a previously described approximation method used for pMHC binding affinity predictions (Lundegaard et al. 2008b). In this benchmark, the AST model was the best performing model, significantly outperforming the strongest of the solo prediction methods, NetMHCcons.
We performed an analysis of FP ratios in the evaluation data and found, at a sensitivity of 90 %, an FP ratio of 1.7 % for NetMHCcons vs. 1.5 % for the AST model. While small, this difference in FP ratio can be translated into an estimate of the reduction in laboratory effort when running epitope discovery experiments. For an epitope screen of 1,000 peptides where a sensitivity of 90 % is desired, 17 peptides would need to be tested when using NetMHCcons. Using the AST model, 15 peptides would need to be tested thus decreasing the number of required peptides by 12 %. This reduction becomes especially meaningful when screening whole proteomes, which may contain hundreds of thousands of unique peptides.
The previous study by Jørgensen et al. (2014) also investigated combining pMHC binding affinity predictions with pMHC stability predictions, as in the AS model described in this study. To our knowledge, this is the first study where pMHC binding affinity and stability predictions have been combined with immunogenicity predictions. We show that integrating these three prediction types leads to a consistent and significant increase in performance for T cell epitope prediction over binding affinity predictions alone. This significant performance increase reinforces the notion that there are factors outside pMHC binding that play a role in CTL activation.
Due to the historical emphasis on pMHC binding affinity data generation, pMHC binding affinity prediction methods currently have the highest predictive performance for T cell epitopes. This observation is also reflected in the integrative models. Here, the relative weight on NetMHCcons was 70–90 % in all cases, suggesting that this method is the strongest epitope predictor of the three independent methods used in this study. This observation was supported when testing the predictive performance of the three methods on an independent evaluation data set. NetMHCcons is trained on a data set consisting of over 100,000 data points, whereas NetMHCstab has a training data set of around 5,500 and the immunogenicity model is trained on just 650 data points. With this training data mismatch in mind, it is no surprise that NetMHCcons is able to contribute the most when combined with the other prediction methods.
However, there is currently room for improvement in pMHC stability and T cell propensity prediction. As more data is generated for these prediction types, it is likely that the weights on these prediction types will change in the combined models. An issue that is making the development of accurate peptide immunogenicity prediction methods difficult is the low number of highly confident non-immunogenic MHC ligands (Calis et al. 2013). The low impact in reporting negative results means there is little interest in thoroughly testing and reporting non-responding peptides. However, in order to develop a strong prediction method, a large amount of positive and negative data is required. We hope more groups will keep this in mind when deciding which data they decide to release in the future.
While the AST model is a significant improvement over NetMHCcons, it is apparent that the absolute gain in predictive performance is relatively small. Two main reasons for this exist. First of all, a large proportion of the epitopes present in the IEDB have been identified using rational epitope discovery approaches including in-silico binding affinity screenings to identify potential epitopes. One such example is the subset of dengue epitopes in the evaluation data set (Weiskopf et al. 2013). These epitopes were identified using an in-silico prescreening where only peptides with high predicted binding affinity were selected. Such a bias towards high affinity will naturally favor affinity-based prediction methods in the evaluation, and as expected the AST method performs on par with NetMHCcons on this data set. Concordantly, one would expect that the performance gain by using the AST method would be higher in situations where the evaluation data were unbiased. Looking at another subset of the data in the evaluation set supports this notion. A large proportion of the Vaccinia epitopes were identified using a prescreening procedure with a tolerant filter for binding affinity, hence imposing a lesser bias in the final set of validated epitopes (Assarsson et al. 2007). As expected, on this data set the performance gain using the AST method compared to NetMHCcons is highly statistically significant (data not shown). Given these considerations, it is hence likely that the performance gain reported in this work by using the AST method is underestimated, and would come out higher in real-life situations where the method is used as a prescreening filter in a pipeline for rational epitope discovery.
Another important issue of epitope characterization and evaluation apparent from this study is the large amount of ill-characterized data contained within the public databases. Here, a large proportion of the reported epitopes do not match the binding motif of the reported restriction element suggesting incorrect assigned restrictions and/or suboptimal identification of the minimal epitope. For example, the 273 epitopes discarded in this study, due to not matching the binding motif of the reported HLA restriction element, had an average predicted binding affinity of 7,550 nM. Only in very few cases (less than 10 %, data not shown) do these epitopes contain nested epitope with high predicted binding affinity, thus strongly suggesting an incorrect assignment of the restriction element for these epitopes.
This study shows that a comprehensive T cell epitope prediction method will very likely need to take multiple factors into account, highlighting the importance in generating novel pMHC stability data and immunogenic and non-immunogenic peptide data. A webserver implementing the final method is available at www.cbs.dtu.dk/services/NetTepi.
References
Alexander J, Sidney J, Southwood S et al (1994) Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity 1:751–761
Assarsson E, Sidney J, Oseroff C et al (2007) A quantitative analysis of the variables affecting the repertoire of T cell specificities recognized after vaccinia virus infection. J Immunol 178:7890–7901
Calis JJ, de Boer RJ, Keşmir C (2012) Degenerate T-cell recognition of peptides on MHC molecules creates large holes in the T-cell repertoire. PLoS Comput Biol 8:e1002412. doi:10.1371/journal.pcbi.1002412
Calis JJA, Maybeno M, Greenbaum JA et al (2013) Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol 9:e1003266. doi:10.1371/journal.pcbi.1003266
Falk K, Rötzschke O, Stevanović S et al (1991) Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351:290–296. doi:10.1038/351290a0
Feltkamp MC, Vierboom MP, Kast WM, Melief CJ (1994) Efficient MHC class I-peptide binding is required but does not ensure MHC class I-restricted immunogenicity. Mol Immunol 31:1391–1401
Frankild S, de Boer RJ, Lund O et al (2008) Amino acid similarity accounts for T cell cross-reactivity and for “holes” in the T cell repertoire. PLoS ONE 3:e1831. doi:10.1371/journal.pone.0001831
Harndahl M, Rasmussen M, Roder G et al (2012) Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur J Immunol 42:1405–1416. doi:10.1002/eji.201141774
Harty JT, Tvinnereim AR, White DW (2000) CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol 18:275–308. doi:10.1146/annurev.immunol.18.1.275
Hoof I, Peters B, Sidney J et al (2009) NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61:1–13. doi:10.1007/s00251-008-0341-z
Huseby ES, White J, Crawford F et al (2005) How the T cell repertoire becomes peptide and MHC specific. Cell 122:247–260. doi:10.1016/j.cell.2005.05.013
Jørgensen KW, Rasmussen M, Buus S, Nielsen M (2014) NetMHCstab—predicting stability of peptide-MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology 141:18–26. doi:10.1111/imm.12160
Karosiene E, Lundegaard C, Lund O, Nielsen M (2012) NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 64:177–186. doi:10.1007/s00251-011-0579-8
Keşmir C, Nussbaum AK, Schild H et al (2002) Prediction of proteasome cleavage motifs by neural networks. Protein Eng 15:287–296
Kim Y, Ponomarenko J, Zhu Z et al (2012) Immune epitope database analysis resource. Nucleic Acids Res 40:W525–W530. doi:10.1093/nar/gks438
Larsen MV, Lundegaard C, Lamberth K et al (2007) Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinforma 8:424. doi:10.1186/1471-2105-8-424
Larsen MV, Lundegaard C, Lamberth K et al (2005) An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol 35:2295–2303. doi:10.1002/eji.200425811
Lee JK, Stewart-Jones G, Dong T et al (2004) T cell cross-reactivity and conformational changes during TCR engagement. J Exp Med 200:1455–1466. doi:10.1084/jem.20041251
Lundegaard C, Lamberth K, Harndahl M et al (2008a) NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res 36:W509–W512. doi:10.1093/nar/gkn202
Lundegaard C, Lund O, Buus S, Nielsen M (2010) Major histocompatibility complex class I binding predictions as a tool in epitope discovery. Immunology 130:309–318. doi:10.1111/j.1365-2567.2010.03300.x
Lundegaard C, Lund O, Nielsen M (2012) Predictions versus high-throughput experiments in T-cell epitope discovery: competition or synergy? Expert Rev Vaccines 11:43–54. doi:10.1586/erv.11.160
Lundegaard C, Lund O, Nielsen M (2008b) Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics 24:1397–1398. doi:10.1093/bioinformatics/btn128
Nielsen M, Lundegaard C, Blicher T et al (2007) NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE 2:e796. doi:10.1371/journal.pone.0000796
Nielsen M, Lundegaard C, Lund O, Keşmir C (2005) The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 57:33–41. doi:10.1007/s00251-005-0781-7
Nielsen M, Lundegaard C, Worning P et al (2003) Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 12:1007–1017. doi:10.1110/ps.0239403
Peters B, Bulik S, Tampe R et al (2003) Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J Immunol 171:1741–1749
Peters B, Sette A (2005) Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinforma 6:132. doi:10.1186/1471-2105-6-132
Rammensee H, Bachmann J, Emmerich NP et al (1999) SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213–219
Rudolph MG, Stanfield RL, Wilson IA (2006) How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol 24:419–466. doi:10.1146/annurev.immunol.23.021704.115658
Stranzl T, Larsen MV, Lundegaard C, Nielsen M (2010) NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics 62:357–368. doi:10.1007/s00251-010-0441-4
Tenzer S, Peters B, Bulik S et al (2005) Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol Life Sci 62:1025–1037. doi:10.1007/s00018-005-4528-2
Tung C-W, Ziehm M, Kämper A et al (2011) POPISK: T-cell reactivity prediction using support vector machines and string kernels. BMC Bioinforma 12:446. doi:10.1186/1471-2105-12-446
Tynan FE, Elhassen D, Purcell AW et al (2005) The immunogenicity of a viral cytotoxic T cell epitope is controlled by its MHC-bound conformation. J Exp Med 202:1249–1260. doi:10.1084/jem.20050864
Vita R, Zarebski L, Greenbaum JA et al (2010) The immune epitope database 2.0. Nucleic Acids Res 38:D854–D862. doi:10.1093/nar/gkp1004
Weiskopf D, Angelo MA, de Azeredo EL et al (2013) Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc Natl Acad Sci U S A 110:E2046–E2053. doi:10.1073/pnas.1305227110
Yewdell JW, Bennink JR (1999) Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol 17:51–88. doi:10.1146/annurev.immunol.17.1.51
Zhang H, Lund O, Nielsen M (2009) The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics 25:1293–1299. doi:10.1093/bioinformatics/btp137
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Institutes of Allergy and Infectious Diseases, National Institutes of Health, and Department of Health and Human Services, under Contract No. HHSN272201200010C. MN is a researcher at the Argentinean national research council (CONICET).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Bar-plot showing the accumulated number of wins for each prediction method as a function of the predicted binding affinity for each epitope in the evaluation data set. (PDF 96 kb)
Rights and permissions
About this article

Cite this article
Trolle, T., Nielsen, M. NetTepi: an integrated method for the prediction of T cell epitopes. Immunogenetics 66, 449–456 (2014). https://doi.org/10.1007/s00251-014-0779-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-014-0779-0