
Abstract
Major histocompatibility complex class II (MHCII) molecules play an important role in cell-mediated immunity. They present specific peptides derived from endosomal proteins for recognition by T helper cells. The identification of peptides that bind to MHCII molecules is therefore of great importance for understanding the nature of immune responses and identifying T cell epitopes for the design of new vaccines and immunotherapies. Given the large number of MHC variants, and the costly experimental procedures needed to evaluate individual peptide–MHC interactions, computational predictions have become particularly attractive as first-line methods in epitope discovery. However, only a few so-called pan-specific prediction methods capable of predicting binding to any MHC molecule with known protein sequence are currently available, and all of them are limited to HLA-DR. Here, we present the first pan-specific method capable of predicting peptide binding to any HLA class II molecule with a defined protein sequence. The method employs a strategy common for HLA-DR, HLA-DP and HLA-DQ molecules to define the peptide-binding MHC environment in terms of a pseudo sequence. This strategy allows the inclusion of new molecules even from other species. The method was evaluated in several benchmarks and demonstrates a significant improvement over molecule-specific methods as well as the ability to predict peptide binding of previously uncharacterised MHCII molecules. To the best of our knowledge, the NetMHCIIpan-3.0 method is the first pan-specific predictor covering all HLA class II molecules with known sequences including HLA-DR, HLA-DP, and HLA-DQ. The NetMHCpan-3.0 method is available at http://www.cbs.dtu.dk/services/NetMHCIIpan-3.0.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Major histocompatibility complex (MHC) molecules play a key role in defining the specificity of the cellular immune system by presenting antigens to the immune system cells. In case of MHC class II molecules, these cells are T helper lymphocytes that recognize peptide–MHC complexes on the surface of antigen-presenting cells. Peptides presented by MHC class II molecules are derived from proteins taken up from the extracellular environment. Whereas a large number of peptides can be generated from pathogenic proteins, only a small part of these trigger an immune response. One of the most important events defining which peptides will trigger an immune response is binding to MHCII molecules expressed by the host (Castellino et al. 1997).
The human MHC locus (in humans called HLA for human leukocyte antigens) is extremely polymorphic and encodes thousands of different HLA class II molecules. Characterising the peptide-binding specificities of all the polymorphic MHC class II molecules is a serious experimental challenge. Therefore, during the last decades, large efforts have been put into the development of in silico methods for predicting peptide-binding affinities to MHC class II molecules. Using thousands of peptide-binding data points, several predictors have been developed and benchmarked (for review, see Nielsen et al. 2010b). One very important subset of these predictors consists of the so-called pan-specific methods that are capable of obtaining accurate predictions for molecules with limited or no binding data (Nielsen et al. 2008, 2010a; Zaitlen et al. 2008; Zhang et al. 2005). For MHC class I prediction, it has been demonstrated that a pan-specific approach can benefit from being trained on cross-loci, and even cross-species, data. That is, the predictive performance for HLA-B locus molecules is improved when including HLA-A locus data in the training of the pan-specific MHC class I binding prediction method (and vice versa), and the overall performance of predictions of HLA molecules is improved when including binding data representing non-human MHC molecules (Hoof et al. 2009). Extending this approach to MHC class II is not a trivial task. Differences in sequence polymorphism and corresponding details in the molecular structures across the different MHC class II loci complicate the development of cross-loci and cross-species training strategies. This, combined with the very limited amount of data available for most MHC class II molecules, has limited the application of pan-specific methods to HLA-DR molecules. The understanding of HLA-DP and HLA-DQ binding specificities is limited to a handful of molecules which have been characterised experimentally, and beyond a few mouse H-2 molecules, to the best of our knowledge, no general MHC class II prediction method is available for non-human primates and other non-human species.
The number of state-of-the-art pan-specific methods for MHC class II molecules available up to date is very limited. The classical MHC class II predictor, TEPITOPE (Sturniolo et al. 1999), uses position-specific scoring matrices derived from experimental data. The method is, however, limited to 51 HLA-DR molecules only. In addition to this, a TEPITOPEpan predictor has been developed (Zhang et al. 2012) by extrapolating from the binding specificities of the molecules characterised by TEPITOPE. The method is based on MHC pocket similarities and is capable of providing predictions for any HLA-DR molecule. The same is achieved by the NetMHCIIpan-2.0 predictor (Nielsen et al. 2010a), which outperforms the TEPITOPEpan method in terms of prediction accuracy (Zhang et al. 2012). The method is based on artificial neural networks and uses an MHC binding pocket pseudo sequence combined with the peptide sequence as an input. Like the TEPITOPEpan method, NetMHCIIpan-2.0 predicts binding for all HLA-DR molecules with a known primary sequence.
In this paper, we present a novel pan-specific predictor capable of predicting binding affinities to all HLA class II molecules. The method is based on artificial neural networks and has been trained on more than 50,000 quantitative peptide-binding measurements covering HLA-DR, HLA-DP, HLA-DQ as well as two murine molecules. Using a panel of benchmark setups, we seek to investigate to what extent the pan-specific method outperforms allele-specific approaches and whether it can obtain accurate predictions even for HLA molecules, which have not been experimentally characterised. Arriving at a “true” pan-specific method enabling prediction of the binding specificity for all HLA-II molecules, we end the analysis by conducting the first global analysis covering all prevalent HLA-II molecules, investigating and quantifying the functional diversity of the molecules encoded at the three HLA-II loci.
Materials and methods
Data sets
Training data used to develop the method consisted of quantitative MHC class II peptide-binding data retrieved from the IEDB database (Vita et al. 2010). In total, the training data set comprises 52,062 data points covering 24 HLA-DR, 5 HLA-DP, 6 HLA-DQ and 2 mouse (H-2) molecules. All molecules were covered by more than 50 peptide binding data points measured as IC50/EC50 values which were log-transformed to fall in the range between 0 and 1 using the relation 1 − log(IC50nM)/log(50,000) (Nielsen et al. 2003). The evaluation set was restricted to HLA-DR molecules and contained 9,860 binding affinity measurements covering 13 molecules, four of which were not included in the training set. A summary of the data used to develop the method is presented in Table S1, and evaluation data set details are given in Table S2.
Mapping of MHC molecules
For constructing the NetMHCIIpan method, all MHC class II molecules need to be mapped to a common reference sequence. This is done by aligning alpha and beta chain sequences of all MHC molecules to the reference sequences, DRA101*01 and DRB101*01. For HLA-DR molecules, the mapping on a sequence level is in agreement with the mapping on the structural level. On the other hand, HLA-DP and HLA-DQ molecules demonstrate minor variations from HLA-DR in the peptide-binding domain in both the alpha and beta chains. To evaluate the structural impact of these variations, we employed the analysis described below. The analysis is based on the five available structures solved for HLA-DP and HLA-DQ molecules, which are compared to a representative high-resolution HLA-DR structure selected among the large number of structures available for HLA-DR molecules. The list of available HLA-DP and HLA-DQ structures from the Protein Data Bank (PDB) is given in Table 1.
An HLA-DR (PDB ID: 1A6A Ghosh et al. 1995) structure was chosen as a reference, and the HLA-DQ and HLA-DP structures were aligned to the binding domain of this reference molecule (Fig. 1). The superimpositions were performed in PyMOL (Schrodinger 2010) and demonstrate a high degree of structural conservation among the different loci (RMSD values between 0.7 and 0.8 Å).

Superimposition of HLA-DR, HLA-DP and HLA-DQ alpha chains. HLA-DR alpha chain (PDB ID: 1A6A Ghosh et al. 1995) is shown in yellow and was used as a reference chain. HLA-DP chain (PDB ID: 3LQZ Dai et al. 2010) is shown in green, HLA-DQ chain without a gap (PDB ID: 1JK8 Lee et al. 2001) is shown in orange and HLA-DQ chain with a gap (PDB ID: 1S9V Kim et al. 2004) is shown in blue. The area affected by the deletion in DQA sequence is circled
During the analysis, an important variation was observed for HLA-DQ molecules only and was investigated in more detail. We observed that sequences belonging to the HLA-DQA1*04, HLA-DQA1*05 and HLA-DQA1*06 serotype groups (e.g. sequences like HLA-DQA1*04:01) display a single amino acid deletion, which from a pure sequence point of view, corresponds to position 53 in HLA-DRA (Robinson et al. 2001). However, this leads to a shift of the preceding residues in the DQA sequences, which now realign with DRA positions 52 and 53. Although the deletion affects the orientation of the short α-helical segment and the loop (residues 45–52) next to the P1 binding pocket (area marked in Fig. 1), these changes have negligible impact on peptide binding, as the reorientation appears to be a localised change, and very few contacts with the peptide are observed within the area. Due to the minor impact of the area discussed above to the binding of the peptide, an automated sequence alignment approach was chosen to identify the deletion in HLA-DQ sequences. Pair-wise sequence alignments were made and visualized using ClustalW (Larkin et al. 2007). Each HLA-DQ sequence was aligned one by one to the reference sequence of HLA-DR. The results are presented in Fig. 2. Figure 2a shows the alignments of HLA-DQ alpha chains with the amino acid deletion, while Fig. 2b demonstrates alignments of HLA-DQ alpha chains with no deletions. The alignments demonstrated that for all the HLA-DQ sequences that have a deletion, the deletion is consistently found in the same place (position 53 in the reference sequence).

Part of sequence alignments of HLA-DQ alpha chains to HLA-DR reference sequence of HLA-DRA1*0101 molecule. a Sequence alignments of HLA-DQ sequences with gaps, b demonstrates the alignment of other HLA-DQ molecules to the same reference sequence. Reference sequence and the position corresponding to the insertion are marked in bold. The alignments were visualized using ClustalW (Larkin et al. 2007)
MHC class II pseudo sequence
For constructing the NetMHCIIpan method, MHC class II molecules were represented by a pseudo sequence consisting of amino acid residues important for peptide binding. Amino acid residues comprising the pseudo sequence were defined as having their side chains pointing towards the peptide and being within 4.0 Å of the peptide-binding core in one or more of the MHC class II structures (including HLA-DR, HLA-DP and HLA-DQ molecules) available in the PDB (www.pdb.org; Berman et al. 2000). The MHC molecules were aligned using the PyMOL molecular viewer (Schrodinger 2010) and interacting residue positions extracted according to the distance criterion. Among the interacting residues, only those found to be polymorphic across the sequences of MHC molecules used for the training of the method were considered. The final pseudo sequence is composed of 15 residues from the alpha chain and 19 residues from the beta chain. The interaction map between the peptide and MHC pseudo sequence is given in Fig. 3.

Interaction map between the peptide and MHC class II pseudo sequence. The columns give the MHC position numbering separately for alpha and beta chains and refer to HLA-DR. The rows show peptide binding core positions. Red squares marking interaction between a particular position of the peptide and MHC define contacts between corresponding two residues
Method
The NetMHCIIpan-3.0 method was implemented as a conventional feed-forward artificial neural network method as described in detail by Nielsen et al. (2010a). The networks were trained using fivefold cross-validation. The data set was split into five groups of peptides based on a common motif clustering as described by Nielsen et al. (2007b). The difference in network architecture from the study presented by Nielsen et al. (2010a) was that network ensembles were trained with 10, 15, 40 and 60 hidden neurons. The BLOSUM50 matrix was used to encode peptide and MHC sequences for the network trainings. Each training was repeated 10 times with different initial configuration values as described in Nielsen et al. (2010a). In total, 40 (4 different numbers of hidden neurons times 10 different random seeds) networks were used for each training/test set combination leading to 200 (5 folds times 40 networks) networks for each molecule.
Leave-one-out setup
In order to assess the predictive performance of the method in the situation where a molecule is not part of the training data, a leave-one-out (LOO) approach was applied. Using LOO, the binding data for the molecule in question were excluded from the training data. Since our data set has a large number of peptides that have been measured for binding to multiple molecules, we also removed peptides common between the evaluation and training data sets to ensure unbiased LOO trainings. In order not to reduce the training set too much in this type of LOO trainings, the evaluation set was split into three subsets resulting into three different fivefold cross-validation trainings for each molecule. The details about such LOO setup are described in Karosiene et al. (2012).
Nearest neighbour approach (NN-finder)
In order to evaluate the performance of the pan-specific method on the molecules that are not found in the training set, we set up a nearest neighbour prediction approach which in this study we call NN-finder. This approach represents the simplest method where the predictions of a query molecule are obtained by first finding its nearest neighbour and using a subsequent allele-specific method to predict the query binding specificity. First of all, for each molecule in question, we found a corresponding nearest neighbour from the training set. The distance between two MHC molecules was calculated from the amino acid similarity between the two pseudo sequences as described by Nielsen et al. (2008), and the nearest neighbour to the molecule in question was defined as the molecule in the training set having the shortest distance. The binding data of each nearest neighbour were then used as training data for the corresponding query molecule. We retrained an allele-specific method from those training data using the NNAlign method (Andreatta et al. 2011) with settings identical to those used for NetMHCII (Nielsen et al. 2007b). The predictive performance for each query molecule was obtained by using its binding data as an evaluation set. In order for the performance to be directly comparable to the LOO results, the splitting of the evaluation set into three subsets was also used here.
Performance measures and statistical analysis
The predictive performance was measured in terms of Pearson's correlation coefficient (PCC) and area under the ROC curve (AUC). PCC values vary between 0 and 1, where 1 represents perfect predictions and 0 random predictions. For AUC measures, a performance value of 1 corresponds to a perfect prediction, and a value of 0.5 reflects random predictions. For more details concerning the performance measures, see Nielsen et al. (2010a). Throughout this study, PCC and AUC values were compared for different methods and evaluated using binomial tests with a significance level of 0.05.
Generation of HLA-II distance trees
For generation of the HLA-II distance tree, the most prevalent alpha and beta chains in the European population were selected as defined by the allele frequencies database (http://www.allelefrequencies.net) (Gonzalez-Galarza et al. 2011). At a frequency threshold of 1 %, we found 21 HLA-DR1, 3 HLA-DPA1, 12 HLA-DPB1, 12 HAL-DQA1 and 13 HLA-DQB1 alleles. We constructed all HLA-DPA1–HLA-DPB1 and HLA-DQA1–HLA-DQB1 combinations arriving at a total of 21 HLA-DR, 36 HLA-DP and 156 HLA-DQ molecules. Sorting (on a per-loci level) the different molecules on descending population frequencies, we constructed a functional redundancy deduced set containing 72 molecules using the Hobohm1 algorithm (Hobohm et al. 1992) with redundancy defined as two molecules sharing a Pearson’s correlation coefficient of 0.99 or above when comparing the predicted binding affinities on a set of 200,000 random natural 15-mer peptides. The set of 72 non-redundant HLA-II molecules is comprised of 21 HLA-DR, 14 HLA-DP and 37 HLA-DQ molecules. Next, we applied the MHCcluster method (Thomsen et al. 2013) to construct a tree describing the functional similarity between the different molecules. In short, the MHCcluster method functions as follows. Binding affinities of a set of 200,000 natural random 15-mer peptides are predicted for each of the HLA molecules using NetMHCIIpan-3.0. Next, the functional similarity between any two HLA molecules is defined by correlating the union of the predicted top 10 % strongest binding peptides for each molecule. The similarity is 1 if the two HLA molecules are predicted to have a perfectly overlapping peptide repertoire and negative if there is no or very limited overlap. The distance between two molecules is defined as 1 − similarity. By using the unweighted pair group method with arithmetic mean clustering, the distance matrix is converted to a distance tree. Generating 100 distance trees using bootstrap estimates the significance of the distance tree. The trees are next summarized, and a consensus tree is made with branch bootstrap values. Sequence logos were constructed from the predicted binding core of the top 1 % strongest predicted binders using Seq2Logo method with default settings (Thomsen and Nielsen 2012).
Results
In the following section, we give the results of applying the new pan-specific method: NetMHCIIpan-3.0 to predict binding for a large set of MHC class II molecules from three human class II loci as well as a small set of mouse H-2 molecules.
NetMHCIIpan-3.0 method’s new approach for getting pseudo sequence
In the most recent pan-specific MHC class II prediction method, NetMHCIIpan-2.0, the pseudo sequence is composed of 21 amino acids from positions within the HLA-DR beta chain that are in potential contact with a peptide using a 4.0 Å distance cut-off and polymorphic across the set of sequenced MHC class II molecules available at the time of the study (Nielsen et al. 2010a). For the NetMHCIIpan-3.0 method described here, the pseudo sequence contains 19 residues from the beta chain of MHC molecules. The main difference between two pseudo sequence obtaining approaches resulting into different number of pseudo sequence positions is that the NetMHCIIpan-3.0 considers polymorphism across the sequences of MHC molecules from the training set only. In order to evaluate this new approach for obtaining the pseudo sequence, we performed a fivefold cross-validation training and compared the results of those reported for NetMHCIIpan-2.0 (Nielsen et al. 2010a). In this comparison, only HLA-DR molecules were considered due to availability of results from both methods. Moreover, for the new method, only the part of the pseudo sequence corresponding to beta chain positions was included as the NetMHCIIpan-2.0 method only includes beta chain residues in the pseudo sequence. The results are shown in Table 2.
The results in Table 2 demonstrate that the new approach for obtaining the pseudo sequence leads to a significantly (p values < 0.05) improved predictive performance compared to the original approach when the pan-specific training approach is applied to the HLA-DR data set. The average increased from 0.688 to 0.695 and from 0.846 to 0.847 for PCC and AUC values, respectively. NetMHCIIpan-3.0 achieves the highest performance for most of the molecules (PCC values are higher for 20 out of 24 molecules, and AUC values are higher for 17 out of 23 molecules, excluding ties). The results demonstrate that the new approach of obtaining pseudo sequences for the neural network trainings improves the predictive performance of the method.
Per-locus training versus cross-loci training
To the best of our knowledge, all pan-specific prediction methods for HLA class II molecules available up to date are limited to HLA-DR. In this study, we introduce an approach for combining residues from the alpha and beta chains into one pseudo sequence. The procedure for the pseudo sequence construction is universal to all MHC class II complexes allowing the pan-specific method to be trained in a cross-loci/cross-species manner (see “Materials and methods”) arriving at one common method suitable for all MHC class II molecules.
To evaluate how such a cross-loci/cross-species impacts the predictive performance of the method, we compared per-locus (and per molecule, see below) training with the pan-specific training including cross-loci data. The results are shown in Fig. 4. Detailed results are given in Table S3. The figure gives average PCC and AUC values for each locus when the method was trained in a cross-loci manner including all HLA molecules and when trained using binding data restricted to each locus, respectively. As can be seen from the figure, the overall performance of the two training approaches is similar. For HLA-DR, the predictive performance improved when training in a cross-loci manner compared to per-locus training. For HLA-DQ and HLA-DP, the performance on the other hand is slightly reduced. This reduction is, however, only significant for HLA-DQ and only when measuring AUC performance values.

Comparison of the method performance when trained on per-locus data and cross-loci data. Average PCC and average AUC values for each locus are demonstrated on the left and right panel, respectively. Significant p values are given above the bars for corresponding loci. The difference in predictive performance between the per-locus and cross-loci training is significant only for HLA-DQ when measuring AUC performance values
Pan-specific versus allele-specific method
As a pan-specific approach, the method presented in this study benefits from the information even from molecules covered by limited binding data or molecules from different loci/species. To demonstrate this, we present a comparison of the performance values obtained for the NetMHCIIpan-3.0 method and allele-specific NN-align method using fivefold cross-validation (see Table 3). The NN-align prediction method was trained as described by Nielsen and Lund (2009), using the same data partitioning based on the common motif clustering approach as used for NetMHCIIpan-3.0.
The results presented in Table 3 demonstrate that the pan-specific NetMHCIIpan-3.0 predictor significantly outperforms the allele-specific NN-align method (p value < 0.0001 for both PCC and AUC values). These results show that the pan-specific method benefits from the binding data measured to different molecules. It also demonstrates that adding data from other molecules significantly boosts the performance for molecules represented by limited peptide-binding measurements. Out of 10 molecules described by less than 400 data points and less than 100 binders, 10 and 9 are shown to obtain higher performance using pan-specific predictor in terms of PCC and AUC values, respectively. The allele-specific NN-align method gives higher PCC and AUC values for three molecules all defined by more than 1,700 peptide-binding data.
Leave-one-out performance
In order to demonstrate how the method performs when predicting binding to novel and uncharacterised molecules, we performed a LOO experiment. In the LOO experiment, a molecule in question was excluded from the training data set, and its binding data acted as an evaluation set. Likewise, all peptides, included in the binding data set of the given molecules, were excluded from the training data in order to avoid biassed overlapping between the evaluation and the training sets. This was done in three rounds by removing one third of the peptides from the evaluation set at a time and performing fivefold cross-validation training in each round. The performance for the query molecule was obtained by combining the predictions of all three evaluation subsets. For the benchmark, we compared the results with the predictive performance of the simple allele-specific approach based on finding the nearest neighbour, NN-finder. For this method, the molecule in question and its binding data were also acting as an evaluation set, while the training data were composed of the peptide binding data of the molecule from the training set having the shortest distance to the molecule in question. The results are depicted in Fig. 5 and presented in detail in Table S4. It is apparent from the results that NetMHCIIpan-3.0 outperforms the NN-finder approach for all loci in terms of average PCC and AUC. Even though we find general improvement when comparing the pan-specific method to the nearest neighbour approach, a significant difference (due to the small number of molecules for each subset) is observed only for HLA-DR molecules (p value < 0.0001). The significance for the mouse allelic locus (H-2) was not assessed due to only two molecules being available.

Leave-one-out results for the NetMHCIIpan-3.0 method in comparison with the NN-finder approach. Average performance measures in terms of PCC and AUC are given in the left and right panel, respectively. Significant p values are given above the bars for corresponding loci (not available for H-2 locus)
The method shows decreased predictive performance with the distance to the nearest neighbour from the training set (Fig. 6). The figure illustrates how the predictive performance of the pan-specific method depends on the distance to the nearest neighbour calculated in terms of pseudo sequence similarities as explained in “Materials and methods”. Regression analysis showed that the performance is decreased significantly with the increasing distance (p value = 0.031, exact permutation test).

Predictive performance of the NetMHCIIpan-3.0 method for the molecules from our data set as a function of distance to the nearest neighbour. The performance was obtained using LOO setup as explained in the “Materials and methods” section. The distance to the nearest neighbour was calculated as described by Nielsen at al. (2008). The solid line represents the least square fit for the data
Independent evaluation of the final NetMHCIIpan-3.0 predictor
For the final evaluation of the pan-specific method common for HLA-DR, HLA-DP, HLA-DQ and mouse molecules, the method was trained using all the available data (52,062 data points) and evaluated on an independent HLA-DR evaluation set containing 9,860 data points. The method was compared with the most recent version of the class II pan-specific predictor NetMHCIIpan-2.0 (Nielsen et al. 2010a). From the results given in Table 4, it is apparent that NetMHCIIpan-3.0 outperforms NetMHCIIpan-2.0 (average PCC is 0.603 compared with 0.586 and average AUC 0.807 compared with 0.802). Although the difference in performances was observed not to be significant, the new pan-specific method shows higher performance for most of the molecules from the evaluation set (NetMHCIIpan-3.0 wins 9 out of 13 and 6 out of 12 times in terms of PCC and AUC measures, respectively).
The NetMHCIIpan-3.0 method presented and benchmarked in this paper was implemented as a web server and is available online at http://www.cbs.dtu.dk/services/NetMHCIIpan-3.0.
Functional clustering of HLA class II molecules
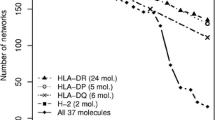
Given the potential of the NetMHCpan-3.0 method to predict binding for any MHC class II molecules with known alpha and beta chain protein sequences, we next applied the method to give an overall estimate of the functional diversity of molecules from the HLA-DR, HLA-DP and HLA-DQ loci molecules. The analysis of the most prevalent alpha and beta chains in the European population was done as described in the “Materials and methods”, and the result is shown in Fig. 7. From the figure, it is apparent that the molecules encoded at the three loci display very limited functional overlap. Also, one can notice that the HLA-DP locus molecules display a very limited functional diversity compared to the HLA-DR and HLA-DQ loci molecules. This is also reflected when measuring the functional diversity of an HLA locus in terms of the mean and standard deviation of the intra-locus distances. Here, we find that the mean intra-distance is significantly shorter (p < 0.001, Student's t test) for HLA-DP compared to HLA-DQ and HLA-DR. We can further relate these differences in functional diversity to the degree of polymorphism at a population level of the HLA pseudo sequences of each locus. Estimating polymorphism in terms of the Kullback–Leibler information content (or divergence sum) (Kullback and Leibler 1951) for the 34 positions in the pseudo sequence for the three loci, we find that this value is significantly higher (p < 0.001, t test) for DP compared to DQ and DR, hence demonstrating that DP molecules share a significantly lower degree of polymorphism compared to the molecules at the two other loci.

Functional clustering of the 72 HLA molecules from the European population. HLA-DR molecules are displayed in red, HLA-DP molecules are displayed in green and HLA-DQ molecules are shown in blue. Sequence logos showing the binding motif are presented for selected molecules representing the different specificity groups
In terms of the predicted functionality, we recover for HLA-DRB1 the overall clustering proposed earlier (Nielsen et al. 2008) with 9 well-defined subgroups (supertypes). For HLA-DQ, the overall functionality seems reduced compared to HLA-DR, with only 5/6 well-defined subgroups, and as stated above, HLA-DP seems to encode for the least functionally diverse set of molecules with only one specificity group being present. Sequence logos for selected subgroups and subgroup representatives are included in the figure to illustrate the functional difference between the different molecules. In general, the predicted binding motifs are in agreement with the motifs proposed earlier for the limited set of HLA class II molecules experimentally characterised by peptide binding data (Andreatta and Nielsen 2012; Andreatta et al. 2011).
Discussion and conclusion
Identification of peptides binding to MHC is a critical step in understanding T cell immune responses. The human MHC genomic region (HLA) is extremely polymorphic comprising several thousands alleles, many encoding a distinct molecule. The potentially unique specificities remain experimentally uncharacterised for the vast majority of HLA molecules.
The sequences of human MHC class II molecules stored in the IMGT database (Robinson et al. 2001) cover over 600 different HLA-DR variants and more than 6,000 different combinations of HLA-DP and HLA-DQ alpha and beta chains. Of these many molecules, less than 30 HLA-DR and only 5 HLA-DP and 6 HLA-DQ molecules have been experimentally characterised with binding data allowing for an accurate estimate of their binding specificity. In order to span this gap, several methods have been developed and benchmarked during the last decade for the prediction of peptide binding to MHC class II molecules (for review, see Nielsen et al. 2010b). Here, pan-specific methods play an important role, as they are capable of giving predictions to those molecules, which have not yet been characterised experimentally. However, until now, MHC class II pan-specific binding prediction approaches have been limited to HLA-DR molecules, leaving a gap in the general understanding of binding specificities for HLA-DP and HLA-DQ molecules (Nielsen et al. 2010b).
In this paper, we present a pan-specific method, NetMHCIIpan-3.0, capable of predicting peptide binding to all HLA molecules. To the best of our knowledge, this is the first predictor common for HLA-DR, HLA-DP and HLA-DQ molecules. The method is based on artificial neural networks and is trained on 52,062 quantitative peptide binding data covering all HLA as well as two mouse molecules.
NetMHCIIpan-3.0 uses a new approach for defining the peptide-binding environment of MHC in terms of pseudo sequence as compared with the most recent NetMHCIIpan-2.0 method (Nielsen et al. 2010a). The main difference between the two approaches for obtaining pseudo sequence is that for NetMHCIIpan-3.0, only polymorphism within the training set is considered whereas the NetMHCIIpan-2.0 method includes polymorphism across all known MHC class II sequences. Our results demonstrated that the new approach for defining the pseudo sequence leads to a significantly improved predictive performance.
Several large-scale benchmarks were carried out that demonstrated that the NetMHCIIpan-3.0 method fulfils the requirements for the pan-specific methods. Its performance was found to be significantly better than that of the allele-specific NN-align predictor (Nielsen and Lund 2009). In particular, the method outperformed NN-align for molecules characterised with only a limited number of binding data. These results hence agree with the results obtained when benchmarking the original NetMHCIIpan method (Nielsen et al. 2010a) and underline the unique power of the pan-specific approach in providing accurate predictions also for molecules characterised with limited peptide-binding data as it has also been demonstrated previously for MHC class I predictions (Karosiene et al. 2012; Nielsen et al. 2007a; Zhang et al. 2009b).
To mimic the situation where the NetMHCIIpan-3.0 method is applied to predict binding for uncharacterised MHC molecules, we conducted a panel of LOO experiments. In these experiments, binding data for one MHC molecule at a time were removed from the training, and the predictive performance next evaluated on the left-out data. The LOO results demonstrated that the proposed method is capable of predicting binding affinity for the molecules for which no binding data are available in the training process. In addition to this, the method showed decreased predictive performance with the distance to the nearest neighbour from the training set, which is in agreement with previous studies on MHC class I (Hoof et al. 2009; Karosiene et al. 2012; Zhang et al. 2009a).
From the results included here, one can notice that HLA-DP molecules demonstrate higher performance values compared with DR and DQ performances. As this high performance is maintained also for the allele-specific (and pseudo sequence independent) NN-align approach, the high performance is not due to the fact that DP molecules are found to be very close to each other in terms of pseudo sequence similarity and function. The reason for this different performance is rather related to the differences within distributions of the binding data available for each locus. The HLA-DP data are very well separated with the majority of the data being either strong or very weak binders. This is in strong contrast to the data for DQ and DR molecules where the majority of the data have intermediate binding affinity (data not shown). This difference in binding affinity distribution strongly influences the predictive performance, as well-separated data sets (as is the case for DP) in general achieve a higher predictive performance. To prove this further, we have performed the analysis where we, in the evaluation of the prediction methods, mimicked the distribution of the HLA-DP data for the DR loci. The analysis demonstrated that the performance for DR molecules is significantly increased when the data match the distribution observed for the DP molecules compared to the original DR data distributions (data not shown).
The NetMHCIIpan-3.0 predictor showed higher performance when compared with the NetMHCIIpan-2.0 method on the external evaluation set. The increased performance of the NetMHCIIpan-3.0 method demonstrates its promising ability to improve when more data become available for molecules from other loci/species.
We further presented a powerful application of the developed pan-specific predictor. We applied the NetMHCIIpan-3.0 method to functionally cluster the most prevalent HLA alleles of the European population. For HLA class I, clustering of molecules into supertypes was proposed by the analysis carried out using experimental data (Lund et al. 2004; Sette and Sidney 1999) and extended by applying pan-specific class I predictor (Nielsen et al. 2007a). However, for MHC class II, the amount of experimental data remains too limited to perform such a cluster analysis, which therefore so far has been limited to HLA-DR molecules (Nielsen et al. 2008). The analysis performed here hence is the first study suggesting reduction of polymorphism of HLA class II molecules by definition of clusters based on similarities in predicted functional binding specificities. Such clustering builds a base for facilitating identification of T helper cell epitopes within different ethnic groups having a high value in the design of epitope-based vaccines.
As we have discussed earlier, developing a cross-loci method for MHC class II is complicated due to the differences of sequences and structures of different loci (Nielsen et al. 2010a). However, as also suggested in this earlier publication, with increasing amounts of binding data covering HLA-DP and HLA-DQ molecules, pan-specific methods may benefit from cross-loci training. In this study, we demonstrated that this is indeed the case. The performance of the proposed NetMHCIIpan-3.0 method when trained on cross-loci was shown to be comparable with that of a method trained on per-loci data. So far, no significant improvement was found between the per-loci and cross-loci trained method. However, this is most likely due to the very low number of HLA-DQ and HLA-DP molecules included in the study and is expected to change with the inclusion of more data covering HLA-DP and HLA-DQ molecules. The situation is hence parallel to that observed for MHC class I. Here, at first, only limited data characterising HLA-A and HLA-B molecules were available for development of the original version of NetMHCpan, and an optimal performance was obtained when the method was trained in a loci-specific manner (Nielsen et al. 2007a). Only when binding data became available covering more HLA molecules as well as MHC molecules from non-human species (including non-human primates) was a cross-loci/cross-species training strategy found to be optimal (Hoof et al. 2009).
The training strategy outlined here for the MHC class II pan-specific prediction method is highly flexible and readily allows inclusion of novel data both in terms of peptides and MHC molecules. This flexibility makes the method a powerful and unique platform for the development of a pan-specific MHC class II predictor covering not only the human class II molecules but also MHC molecules from other species of interest. Lessons learned from MHC class I suggest that such a “true” pan-specific approach is feasible and that prediction accuracies for both human and non-human MHC molecules can be greatly boosted given the ability of the pan-specific method to leverage information across species and loci (Nene et al. 2012).
In conclusion, we believe the proposed NetMHCIIpan-3.0 method is an important step forward in boosting MHC class II binding predictions covering a large number of molecules from different species and therefore reduces experimental costs for the immunologists working within the field of epitope-based vaccine design.
References
Andreatta M, Nielsen M (2012) Characterizing the binding motifs of 11 common human HLA-DP and HLA-DQ molecules using NNAlign. Immunology 136(3):306–311. doi:10.1111/j.1365-2567.2012.03579.x
Andreatta M, Schafer-Nielsen C, Lund O, Buus S, Nielsen M (2011) NNAlign: a web-based prediction method allowing non-expert end-user discovery of sequence motifs in quantitative peptide data. PLoS One 6(11):e26781. doi:10.1371/journal.pone.0026781
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28(1):235–242
Castellino F, Zhong G, Germain RN (1997) Antigen presentation by MHC class II molecules: invariant chain function, protein trafficking, and the molecular basis of diverse determinant capture. Hum Immunol 54(2):159–169
Dai S, Murphy GA, Crawford F, Mack DG, Falta MT, Marrack P, Kappler JW, Fontenot AP (2010) Crystal structure of HLA-DP2 and implications for chronic beryllium disease. Proc Natl Acad Sci U S A 107(16):7425–7430
Ghosh P, Amaya M, Mellins E, Wiley DC (1995) The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature 378(6556):457–462. doi:10.1038/378457a0
Gonzalez-Galarza FF, Christmas S, Middleton D, Jones AR (2011) Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res 39(Database issue):D913–D919
Hobohm U, Scharf M, Schneider R, Sander C (1992) Selection of representative protein data sets. Protein Sci 1(3):409–417. doi:10.1002/pro.5560010313
Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M (2009) NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61(1):1–13. doi:10.1007/s00251-008-0341-z
Karosiene E, Lundegaard C, Lund O, Nielsen M (2012) NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 64(3):177–186. doi:10.1007/s00251-011-0579-8
Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM (2004) Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci U S A 101(12):4175–4179. doi:10.1073/pnas.0306885101
Kullback S, Leibler RA (1951) On information and sufficiency. Ann Math Stat 22(1):142–143
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948
Lee KH, Wucherpfennig KW, Wiley DC (2001) Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat Immunol 2(6):501–507. doi:10.1038/88694
Lund O, Nielsen M, Kesmir C, Petersen AG, Lundegaard C, Worning P, Sylvester-Hvid C, Lamberth K, Roder G, Justesen S, Buus S, Brunak S (2004) Definition of supertypes for HLA molecules using clustering of specificity matrices. Immunogenetics 55(12):797–810. doi:10.1007/s00251-004-0647-4
Nene V, Svitek N, Toye P, Golde WT, Barlow J, Harndahl M, Buus S, Nielsen M (2012) Designing bovine T cell vaccines via reverse immunology. Ticks Tick Borne Dis 3(3):188–192
Nielsen M, Justesen S, Lund O, Lundegaard C, Buus S (2010a) NetMHCIIpan-2.0—improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res 6:9
Nielsen M, Lund O (2009) NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinforma 10:296
Nielsen M, Lund O, Buus S, Lundegaard C (2010b) MHC class II epitope predictive algorithms. Immunology 130(3):319–328. doi:10.1111/j.1365-2567.2010.03268.x
Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O, Buus S (2007a) NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One 2(8):e796. doi:10.1371/journal.pone.0000796
Nielsen M, Lundegaard C, Blicher T, Peters B, Sette A, Justesen S, Buus S, Lund O (2008) Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput Biol 4(7):e1000107. doi:10.1371/journal.pcbi.1000107
Nielsen M, Lundegaard C, Lund O (2007b) Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinforma 8:238
Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O (2003) Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 12(5):1007–1017. doi:10.1110/ps.0239403
Robinson J, Waller MJ, Parham P, Bodmer JG, Marsh SG (2001) IMGT/HLA Database—a sequence database for the human major histocompatibility complex. Nucleic Acids Res 29(1):210–213
Schrodinger, LLC (2010) The PyMOL molecular graphics system, version 1.3r1
Sette A, Sidney J (1999) Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics 50(3–4):201–212
Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, Braxenthaler M, Gallazzi F, Protti MP, Sinigaglia F, Hammer J (1999) Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol 17(6):555–561. doi:10.1038/9858
Thomsen M, Lundegaard C, Buus S, Lund O, Nielsen M (2013) MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics. doi:10.1007/s00251-013-0714-9
Thomsen MC, Nielsen M (2012) Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res 40(Web Server issue):W281–W287
Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B (2010) The immune epitope database 2.0. Nucleic Acids Res 38(Database issue):D854–D862
Zaitlen N, Reyes-Gomez M, Heckerman D, Jojic N (2008) Shift-invariant adaptive double threading: learning MHC II-peptide binding. J Comput Biol 15(7):927–942. doi:10.1089/cmb.2007.0183
Zhang GL, Khan AM, Srinivasan KN, August JT, Brusic V (2005) MULTIPRED: a computational system for prediction of promiscuous HLA binding peptides. Nucleic Acids Res 33(Web Server issue):W172–W179
Zhang H, Lund O, Nielsen M (2009a) The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics 25(10):1293–1299
Zhang H, Lundegaard C, Nielsen M (2009b) Pan-specific MHC class I predictors: a benchmark of HLA class I pan-specific prediction methods. Bioinformatics 25(1):83–89. doi:10.1093/bioinformatics/btn579
Zhang L, Chen Y, Wong HS, Zhou S, Mamitsuka H, Zhu S (2012) TEPITOPEpan: extending TEPITOPE for peptide binding prediction covering over 700 HLA-DR molecules. PLoS One 7(2):e30483. doi:10.1371/journal.pone.0030483
Acknowledgments
MN is a researcher at the Argentinean national research council (CONICET). This project has been funded in whole or in part with federal funds from the National Institutes of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract nos. HHSN272201200010C and HHSN272200900045C.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 203 kb)
Rights and permissions
About this article
Cite this article
Karosiene, E., Rasmussen, M., Blicher, T. et al. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 65, 711–724 (2013). https://doi.org/10.1007/s00251-013-0720-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-013-0720-y