
Abstract
The generation and maintenance of allelic polymorphism in genes of the major histocompatibility complex (MHC) is a central issue in evolutionary genetics. Recently, the focus has changed from ex situ to in situ populations to understand the mechanisms that determine adaptive MHC polymorphism under natural selection. Birth-and-death evolution and gene conversion events are considered to generate sequence diversity in MHC genes, which subsequently is maintained by balancing selection through parasites. The ongoing arms race between the host and parasites leads to an adaptive selection pressure upon the MHC, evident in high rates of non-synonymous vs synonymous substitution rates. We characterised the MHC class II DRB exon 2 of free living bank voles, Clethrionomys glareolus by single-strand conformation polymorphism and direct sequencing. Unlike other arvicolid species, the DRB locus of the bank vole is at least quadruplicated. No evidence for gene conversion events in the Clgl–DRB sequences was observed. We found not only high allelic polymorphism with 26 alleles in 36 individuals but also high rates of silent polymorphism. Exceptional for MHC class II genes is a purifying selection pressure upon the majority of MHC–DRB sequences. Further, we analysed the association between certain DRB alleles and the parasite burden with gastrointestinal trichostrongyle nematodes Heligmosomum mixtum and Heligmosomoides glareoli and found significant quality differences between specific alleles with respect to infection intensity. Our findings suggest a snapshot in an evolutionary process of ongoing birth-and-death evolution. One allele cluster has lost its function and is already silenced, another is loosing its adaptive value in terms of gastrointestinal nematode resistance, while a third group of alleles indicates all signs of classical functional MHC alleles.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
From an evolutionary point of view, the genes of the major histocompatibility complex (MHC) are of a special interest because they contain some of the most polymorphic coding genes in the vertebrate genome (Bernatchez and Landry 2003). The MHC comprises a multi-gene family coding for transmembrane receptor glycoproteins. These MHC molecules bind antigens and present them to T lymphocytes that initiate the following immune answer by recognising a foreign peptide (Brown et al. 1993). The MHC can be divided in two major groups. Class I molecules mostly are responsible for the recognition and binding of intracellular antigens, whereas class II molecules correspond to extracellular antigens (Klein and Sato 1998). Due to its important role in the vertebrate immune system, it is assumed that the extraordinary high level of polymorphism is maintained by balancing selection driven by pathogens and parasites (Apanius et al. 1997; Bernatchez and Landry 2003; Hedrick 1999, 2002; Stear et al. 2005). Thereby, operating evolutionary mechanisms are of great interest and subject to ongoing controversy (Bernatchez and Landry 2003; Hedrick 1999; Piertney and Oliver 2006; Sommer 2005). High rates of non-synonymous (d N) vs synonymous (d S) substitutions are regarded to be good indicators for an ongoing adaptive evolutionary process (Parham and Otha 1996). This prediction has been proven in a number of recent field studies under natural selection conditions and was mostly combined with high levels of allelic polymorphism (Bryja et al. 2006; Froeschke and Sommer 2005; Harf and Sommer 2005; Meyer-Lucht and Sommer 2005; Oliver and Piertney 2006; Schad et al. 2004, 2005). It holds especially true for certain nucleotide positions defined as antigen-binding sites (ABS) based on the human sequence (Brown et al. 1993). These are key positions in the peptide-binding region of the MHC molecule where a single substitution can change the binding properties of the molecule radically. Moreover, it becomes more and more evident that not only allelic diversity within one single locus is important. Gene duplication seems to be an important evolutionary tool to increase allelic diversity within a species and, thereby, its adaptive potential to new challenges. Generation of new alleles can be achieved via gene conversion events (segmental exchanges of sequence motives between alleles of the same or different loci) or via birth-and-death evolution (Nei et al. 1997; Parham and Otha 1996). Birth-and-death evolution produces new orthologous genes by duplication that diverge by accumulating mutations over time. Some of these alleles persist in the genome, and some get deleted or become pseudogenes (Nei and Rooney 2005). Duplicated MHC genes can be found in a broad range of free-ranging animals (e.g. Baker et al. 2006; Bryja et al. 2006; Harf and Sommer 2005; Miller and Lambert 2004; Miska et al. 2004; Phillips et al. 2003; Reusch et al. 2004; Schwensow et al. 2007) and strengthen the assumption that gene duplication is an important process in MHC evolution.
To sharpen our understanding of the ongoing evolutionary mechanisms that determine adaptive MHC polymorphism, it is necessary to investigate free-living populations evolving under natural selection pressures (Bernatchez and Landry 2003). For this purpose, we chose the bank vole, Clethrionomys glareolus, a common European Muridae belonging to the subfamily Arvicolinae (Michaux et al. 2001). C. glareolus has been subject to a broad range of scientific studies including parasitological, sociobiological, reproductive and population studies making it an interesting model species for further studies (e.g. Barnard et al. 2003; Behnke et al. 2001; Bujalska and Hansson 2000; Haukisalmi and Henttonen 1993a,b, 2000; Horne and Ylönen 1996; Hörnfeldt 2004; Norrdahl and Korpimäki 2002; Ylönen et al. 1997). Our specific goals were (1) to investigate the variation of the MHC class II DRB locus, (2) to analyse selection, expression and evolution patterns and finally, (3) to compare these data with infection data of gastrointestinal helminths.
Materials and methods
We life-trapped 130 individuals of bank voles (C. glareolus) from the end of June to the beginning of September 2004 in a deciduous forest about 35 km north east of Hamburg, Germany. Ear tissue samples were taken and stored in 70% ethanol at 5°C. Individual faecal samples were collected from each trap and fixed in 70% ethanol to assess the individual intestinal parasite burden. Traps were cleaned before re-use.
Genetic investigations focuses on the MHC class II DRB gene exon 2 because it contains most of the functional important ABS and is, therefore, the most likely candidate for detecting balancing selection acting on MHC class II genes. We analysed a subsample of 36 individuals using polymerase chain reaction (PCR) with two different primer systems (PS1, PS2), single-strand conformation polymorphism (SSCP) and direct sequencing. Genomic DNA was extracted using the DNeasy™ Tissue Kit (Qiagen®, Hilden) following the manufacturer’s protocol. PS1 used the primers Tub1JS (Schad et al. 2004) and YML10 (Meyer-Lucht & Sommer, unpublished) to amplify a fragment of 195 bp, while PS2 used the new designed forward primer JAFW1 (5′-GTGCGGTTTCTGGACAGATAC-3′) and YML10 to amplify 167 bp. All PCRs were performed in a reaction volume of 20 μl, each containing 40–100 ng of DNA, 0.375 mM of each primer (Sigma Aldrich®, St Louis), of 10× reaction buffer, 0.175 mM deoxyribonucleotide triphosphates, 1U Taq polymerase (all Quantum Appligene®). Thermocycling was carried out on a TGradient Thermocycler (Biometra, Göttingen) with an initial denaturation step 120 s at 96°C followed by 35 cycles of 30 s denaturation at 96°C, 60 s annealing at 59°C for PS1 and at 57°C for PS2, 60 s elongation at 72°C, finished with a 10-min elongation at 72°C and stopped by cooling down to 5°C.
PCR products showed a distinct single band on a 1.5% agarose gel stained with ethidium bromide. After amplification, SSCP on a 15% polyacrylamide gel was conducted based on the manufacturer’s protocol (ETC, Kirchintellinsfurt) and modified for this study. Two to four microlitres of PCR product was mixed with twice the volume of denaturating loading dye (50 mM NaOH + 1 mM ethylenediaminetetraacetic acid, Xylencyanol), heated at 50°C for 10 min, and 6 μl was loaded on the polyacrylamide gel. Electrophoresis was run on a horizontal cooling electrophoresis system (Amersham Pharmacia, Freiburg) at 12°C. Conditions were 20 min at 200 V, 10 mA, 10 W followed by 4.5 h at 450 V, 30 mA and 20 W. Gels were fixed and silver stained (PlusOne DNA Silver Staining Kit, Amersham Pharmacia). Distinct bands were excised from the gel, resolved in 30 μl 1× TBE and incubated for 3 h. Reamplification of single strands was performed as described before with 30 instead of 35 cycles. PCR products were purified by gel extraction (QIAquick, Gelextraction Kit, Qiagen) and ethanol precipitation. Approximately 60–80 ng of DNA was used for cycle sequencing, which was performed using a dye terminator sequencing kit (BigDye® Terminator v3.1, Applied Biosystems, Foster City) and analysed by an automated sequencer (Model 3100, Applied Biosystems). Sequences were edited by eye and aligned in the alignment editor embedded in MEGA 3.1 (Kumar et al. 2004). All alleles were defined on the basis of at least three independent sequences. A basic local alignment search tool (BLAST) search at GenBank (National Centre for Biotechnology Information, NCBI) confirmed gene identity for all Clethrionomys sequences.
RNA (Qiagen RNeasy kit, Qiagen) from fresh liver tissue from two individuals was extracted to determine if DRB alleles that could be grouped to different phylogenetic clusters (see below) are expressed. Genomic DNA was removed via the DNase Set (Qiagen), and first-strand complementary DNA (cDNA) synthesis was performed in a 20-μl reaction tube using SuperScript® III reverse transcriptase (Invitrogen, Karlsruhe) and oligo(dT)17 primer. Incubation was conducted at 55°C for 45 min followed by 15-min step at 70°C. We performed PS1 and PS2 with the obtained cDNA as a template followed by SSCP and direct sequencing.
MEGA 3.1 was used to calculate the relative rates of non-synonymous (d N) and synonymous (d S) substitutions according to the method of Nei and Gojobori (1986) with Jukes and Cantor (1969) correction for multiple hits for all alleles except pseudogenes. The substitution rates were calculated separately for non-antigen-binding sites (nABS) and 19 ABS defined by Brown et al. (1993). The probability that ω (d N/d S)=1 was determined using a t-test. MEGA 3.1 was employed to calculate mean genetic distance (d) based on nucleotide divergence according to the Kimura two-parameter distance. This method was also applied to construct a neighbour-joining phylogenetic tree based on the shared sequence sections of all alleles. A bootstrap analysis with 5,000 replicates was conducted to proof reliability of the branching. Test for recombination events between alleles was conducted with the program Geneconv 1.81 (Sawyer 1999). The program runs 10,000 permutations of the original data to test global and pairwise for the occurrence of possible inner and outer fragments involved in recombination or gene conversion events.
Parasite load was assessed in all genetically investigated individuals (except for one) by a McMaster floatation technique modified by Meyer-Lucht and Sommer (2005). The faecal egg count (FEC), measured in eggs per gram faeces, is a non-invasive technique and has proved to give adequate information about the parasite load of an animal (Froeschke and Sommer 2005; Harf and Sommer 2005; Meyer-Lucht and Sommer 2005; Schad et al. 2005). Helminth eggs were assigned to different morphotypes defined by size and morphological characters. Photographs of all eggs were taken and used for later classification together with adult worms obtained from dissections of dead found animals. We used only the data of the most abundant helminths, the trichostrongyle nematode species Heligmosomum mixtum and Heligmosomoides glareoli, which were observed in more than 96% of the animals tested positive on helminth infection (Axtner and Sommer, unpublished data). The non-parametric Kruskal–Wallis was applied to investigate differences in the parasite load between the most common expressed alleles (present >5 individuals) followed by pairwise Mann–Whitney U-tests. Bonferroni-corrected significance levels were used for multiple comparisons (Rice 1989; Sachs 1992). All tests were performed in SPSS® version 11.5.
Results
MHC sequence variation, selection and phylogenetic relationships
Both primer systems (PS1, PS2) were successfully performed on all 36 individuals revealing 26 different alleles for the DRB exon II of the bank vole. We followed the nomenclature of Klein et al. (1990) and labelled them Clgl DRB*01 to Clgl DRB*28 eliding the numbers 2 and 9 (GenBank accession numbers: EF434791 to EF434816). BLAST search confirmed the homology to other rodent DRB sequences for all alleles (84 to 94% concordance). Thirteen alleles could only be detected with PS1, nine alleles could only be detected with PS2, four alleles could be detected with both primer systems. The maximum number of alleles in a single individual was eight; therefore, the MHC DRB locus must be at least quadruplicated. We could not find any evidence for genetic recombination or gene conversion events between aligned sequences and/or ancestral relicts of such events using the program Geneconv 1.81.
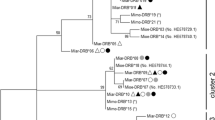
Phylogenetic analysis of the Clgl sequences revealed two distinct monophyletic allele clusters (cluster I and II) each consisting of alleles that could only be amplified by one of the primer systems (Fig. 1). Cluster I comprised the alleles only amplified by PS1 and cluster II the alleles only amplified by PS2. The alleles DRB*16, *17, *18 and *21 did not belong to the monophyletic clusters I and II. We did not combine these alleles to a cluster III because phylogenetic analyses (see below) suggest that they might belong to different lineages. Instead, we analysed them as a separate group (group III) based on shared attributes of classical functional DRB alleles (see below, Fig. 1). Apparently, there was a strong linkage between the alleles Clgl DRB*01 (cluster I), DRB*21 (group III), and DRB*26 (cluster II). In only 2 out of 17 cases, 1 of the 3 alleles was found alone. Due to of the lack of knowledge about the affiliation of the alleles to different loci, it was impossible to test the linkage disequilibrium. Clgl DRB*22 and *25 (cluster II; Fig. 1) seemed to be pseudogenes with stop codons in their sequences and were excluded from further analysis.

Neighbour-joining phylogeny for all 26 C. glareolus alleles using a human sequence as an outgroup (GenBank accession no. AM109973). The scale bar represents the genetic distance and the numbers are bootstrap values > 50 (5,000 replicates). Cluster I was only amplified by PS1, and cluster II was only amplified by PS2, the alleles of group III were amplified by both primer systems
By sequencing the cDNA samples, the alleles Clgl DRB*17, *19 and *24 were found that confirms the expression of at least some cluster II alleles as well as group III alleles. PS1 could not successfully be performed on the cDNA samples suggesting that the cluster I is no longer expressed.
Within the shared part of the 24 Cluster I, II and group III alleles, 70 (42%) nucleotide positions are variable. Separate analysis by allele clusters revealed that one to eight nucleotide positions were polymorphic in cluster I \( {\left( {\overline{x} = 4.87,\,SE = 1.17} \right)} \), 1 to 5 in cluster II \( {\left( {\overline{x} = 3.24,\,SE = 1.26} \right)} \) and 11 to 28 in group III \( {\left( {\overline{x} = 23.33,\,SE = 3.05} \right)} \). Group III showed a five times higher mean genetic distance between its alleles than both clusters I and II (d Cl I = 0.026 ± 0.007; d Cl II = 0.026 ± 0.009; d Gr III = 0.132 ± 0.020). The 24 alleles translate into only 15 different amino acid sequences. In cluster I, the alleles Clgl DRB*03, *05, *13 and Clgl DRB*08, *10, *15 as well as Clgl DRB*12, *14 display the same amino acid patterns, respectively. In the cluster II, Clgl DRB*19, *23, *26 and Clgl DRB*24, *28 show no difference in the amino acid sequence as well as the alleles Clgl DRB*20, *27.
This high level of synonymous substitutions leads in both monophyletic clusters I and II to a ω significantly smaller than the one in analysis over all sites (Table 1). Especially in cluster I, there seemed to be a strong detectable negative selection pressure upon the alleles producing a ω Cl I being 20 times smaller than the ω of cluster II. Within cluster I, there was no difference between the ω at the ABS and the non-ABS, both showing also strong negative selection pressure. For cluster II, d N at the ABS was 0, turning ω consequentially to 0 and, thus, impossible to test vs d S. Only in group III, d N was larger than d S at the ABS although not significant. At the non-ABS, d N was smaller than d S but also not significant. These results in C. glareolus contradict previous findings in free-ranging mammal species under natural selection conditions (Fig. 2).

Differences between the rates of non-synonymous (d N) and synonymous (d S) substitutions (d N–d S) in ABS (shaded bars) and nABS (black bars) of MHC class II DRB exon 2 alleles. The amino acid sequence changing substitution rate of different primate (Microcebus murinus, Microcebus berthae), rodent (A. sylvaticus, A. flavicollis, Leopoldamys sabanus, G. paeba, R. pumilio, Rattus rattus, Hypogeomys antimena), perissodactyla (Equus przewalskii) and artiodactyla (Oryx leucoryx, Damaliscus pygargus pygargus) species (modified from Sommer 2005) is compared to C. glareolus allele clusters I, II and group III (this study)
A phylogenetic analysis with several rodent MHC-DRB sequences emphasised the uniqueness of both allele clusters I and II supported by high bootstrap values (Fig. 3). Cluster I grouped next to a sequence of Rattus norvegicus but showed a great genetic distance to this sequence. Likewise, the alleles of cluster II showed a great distance to their nearest grouping sequences (group III: Clgl DRB*17 and *18). The two alleles Clgl DRB*16 and *21 (group III) showed comparable genetic distances to other observed rodent alleles and grouped with different species that is known as trans-species evolution (Klein 1987).

Neighbour-joining phylogeny of all identified C. glareolus alleles (bold letters) in comparison with 30 DRB sequences of 10 different rodent species, using a human sequence as an outgroup. GenBank accession no. are provided in parentheses. The scale bar represents the genetic distance and the numbers are bootstrap values > 50 (5,000 replicates)
Expressed MHC-alleles and gastrointestinal nematode infection
The adaptive value of expressed MHC alleles was examined by investigations of the gastrointestinal nematode load. For statistical reasons, only expressed alleles of cluster II and group III that were found at least five times were taken into account. Alleles that were coding for the identical amino acid sequence were combined. Specific alleles had a significant influence upon the intensity of infection with trichostrongyle nematodes (df = 4; p = 0.041; Fig. 4). The allele group Clgl DRB*20/*27 (cluster II) seemed to be associated with high susceptibility to gastrointestinal trichostrongyle nematodes, whereas the allele group Clgl DRB*19/*23/*26 (cluster II) and the allele Clgl DRB*21 (group III) are associated with high resistance to these nematodes. Pairwise comparisons revealed significant differences between the alleles Clgl DRB*20/*27 and Clgl DRB*19/*23/*26 (Z = −2.585, p = 0.011; Bonferroni not significant) and between the alleles Clgl DRB*20/*27 and Clgl DRB*21 (Z = −2.391, p = 0.019; Bonferroni not significant; Fig. 4).

Association between specific DRB alleles and gastrointestinal trichostrongyle nematode load (nematode eggs per gram faeces, epg) in C. glareolus. See text for details
Discussion
In the present study, we analysed for the first time the allelic diversity of the MHC class II DRB exon 2 of the bank vole C. glareolus. So far, the only other arvicolid species examined for the DRB locus is the water vole Arvicola terrestris (Oliver and Piertney 2006). Interestingly, the authors observed a very low allelic diversity in the water vole with only five different alleles in 100 individuals sampled from four distinct populations by using the identical genetic approach (SSCP and cycle sequencing). Usually, a low diversity at the MHC is considered as the result of a severe population bottleneck (e.g. Amills et al. 2004; Ellegren et al. 1993; Smulders et al. 2003; Sommer 2003). In other field studies on the DRB of rodents, far more alleles were revealed, for example, 27 alleles in 146 Apodemus flavicollis individuals (Meyer-Lucht and Sommer 2005) and 38 different alleles in 119 individuals of its sister species Apodemus sylvaticus (Musolf et al. 2004), 34 alleles in 40 individuals of Gerbillurus paeba (Harf and Sommer 2005) or 20 alleles in 58 individuals of Rhabdomys pumilio (Froeschke and Sommer 2005). In the present study, we also observed a much higher number of alleles in the bank vole that is more comparable to the former studies mentioned above than to A. terrestris. However, neither of these studies took the expression level of the counted alleles into account. Considering the functionality of MHC-DRB alleles, we observed a low number of classical MHC DRB alleles in our bank vole population, which probably did not experience a severe bottleneck event.
In this context, of high interest is the fact that the DRB locus of C. glareolus is at least quadruplicated. Neither in A. terrestris nor in Peromyscus belonging to an arvicolid sister group, a duplication of the DRB locus has been found (Oliver and Piertney 2006; Richman et al. 2002). However, in C. glareolus as well as in the two vole species A. terrestris and Microtus arvalis, also the MHC class II gene DQA is duplicated (Bryja et al. 2006). This suggests that the duplication took place before the vole species separated. Thus, we can conclude that the quadruplication of the DRB locus in the bank vole is younger on an evolutionary time scale than the duplication of the DQA locus. It occurred at least after the separation of A. terrestris and C. glareolus because it was not found in the former one. The low divergence within the alleles of clusters I and II also indicates that the quadruplication of the DRB locus is a very recent event. Gene duplication is regarded as an important mechanism generating MHC diversity (Gu and Nei 1999; Klein et al. 1998) and was found in a broad range of taxa (e.g. Baker et al. 2006; Bryja et al. 2006; Harf and Sommer 2005; Miller and Lambert 2004; Miska et al. 2004; Phillips et al. 2003; Reusch et al. 2004; Schwensow et al. 2007). Two major theories have been put forward to explain the evolution of the MHC. One hypothesis is the birth-and-death model of evolution (Nei et al. 1997), which suggests that MHC genes are produced by duplications. Some duplicated genes diverge functionally through accumulation of mutations over time, some alleles become fixed, some pseudogenes due to deleterious mutations are deleted from the genome. An alternative hypothesis is gene conversion characterised by a segmental exchange between alleles of one or of different loci leading to new alleles (Parham and Otha 1996). Both hypotheses do not mutually exclude each other, but their relative importance is controversially discussed (reviewed by Martinsohn et al. 1999; Nei and Rooney 2005). In contrast to the evidence for gene conversion events in the sequence data of free living species (e.g. Go et al. 2003; Miller and Lambert 2004; Reusch and Langfors 2005; Richman et al. 2003; Schaschl et al. 2005), we could not find any significant signs for recombination or gene conversion events in the bank vole C. glareolus.
The uniqueness of both C. glareolus clusters I and II is confirmed by phylogenetic analysis with several other rodent DRB sequences included where these cluster turned out to be monophyletic. Nevertheless, BLAST search at GenBank confirmed their homology to the DRB. It seems that the alleles of the cluster I are no longer expressed because PS1 was not successfully performed on the cDNA samples, while at least some alleles of the cluster II and group III were amplified and thus expressed. The phylogenetic analysis revealed that group III alleles belong to different lineages, which provides further evidence that group III alleles are under balancing selection pressure and follow the trans-species mode of evolution. Allelic lineages are maintained over long periods of time, even across speciation events. This has been observed in many genera including rodents (Bryja et al. 2006; Edwards et al. 1997; Musolf et al. 2004; Seddon and Baverstock 2000). Group III alleles grouped next to other rodent species in comparative phylogenetic analysis also indicate that they are older than the separation of the respective rodent taxa.
As already pointed out, gene duplication is a common feature of the MHC genes, and duplicated MHC loci usually are governed by diversifying selection over short evolutionary time, which may favour their specialisation (Hughes 1999). In classical mammalian MHC genes, substitutions in the ABS codons are predominantly replacement substitutions, and therefore, ω (d N/d S) is greater than one indicating balancing selection (Hughes and Nei 1988, 1989, reviewed by Sommer 2005). However, duplicated genes might experience a change or even a loss in function (Nei et al. 1997) and a change in expression divergence (Li et al. 2005). Once the proteins of duplicated genes have become specialised for distinct functions, new amino acid changes are no longer advantageous, and purifying selection might predominate (Hughes and Friedman 2004). In other words, the new generated locus could have undergone a sub- or neofunctionalisation, which imposes it to a new purifying selection pressure (Zhang 2003, Nei 2005). In this case, the number of synonymous substitutions might exceed the non-synonymous rate (d S > d N), resulting in an apparently MHC atypical ω ratio < 1 at the ABS. However, if the locus would have lost its function and became a pseudogene, we would expect a ω ≈ 1 as predicted by neutral theory due to missing selection pressure upon these alleles and a high nucleotide substitution rate, which should be more or less equal to the total mutation rate (Nei 2005).
We investigated evidence for selection on MHC class II DRB in C. glareolus by comparing the non-synonymous to synonymous substitution ratios (ω). In C. glareolus, as expected, investigations of DQA sequences indicated strong positive selection (ω > 1; Bryja et al. 2006). Even in A. terrestris with only five revealed DRB sequences, a positive selection pressure could be observed by means of a ω > 1 (Oliver and Piertney 2006). Contrarily, our study revealed a strong negative selection pressure (ω ≪ 1) upon the majority of MHC DRB sequences (clusters I and II), which suggests purifying selection. Only the DRB sequences of group III followed the expected pattern, although ω was not significantly larger than one probably due to the small number of alleles in this group. As mentioned above, especially in MHC class II genes, it is very uncommon to find a ω smaller than one indicating a negative or purifying selection pressure. Thus, the strong negative selective force especially upon the non-expressed cluster I is quite puzzling. To the best of our knowledge, so far, there is only one study by Jarvi et al. (2004) in Hawaiian honeycreepers (Drepanidinae) revealing a ω < 1. Like our study, the authors found an allele cluster being under negative selection (ω Drep. II = 0.29) and another one being under positive or adaptive selection with a ω larger than one (ω Drep. I = 2.38). We hope that ongoing studies investigating expression divergence in relation to parasites will shed light upon this puzzle.
The pressure exerted by pathogens is considered as the major selection component influencing MHC polymorphism. Several recent studies confirmed the functional significance of MHC class II DRB polymorphism in free-ranging animal population (reviewed by Sommer 2005). The most common alleles of the expressed cluster II and group III were used to analyse their association with parasite burden and their adaptive value. We focused on the most prevalent trichostrongyle nematodes, H. mixtum and H. glareoli, which amount for more than 96% of all helminth infections (Axtner and Sommer, unpublished data) and are, therefore, the most likely candidates to detect co-evolutionary associations. The negative impact of H. mixtum on C. glareolus has been shown ex situ by an increased infection and mortality rate under poor quality food and high transmission efficiency (Haukisalmi and Henttonen 2000). Common parasite species often exhibit high host specificity and are supposed to have undergone a close co-evolution with their host. Contrarily, rare species often have a variety of different host species, thus are less prevalent than common ones and are, therefore, less likely to fuel an arms race between host and parasite (Haukisalmi and Henttonen 2000). In our study population, the allele group Clgl DRB*20/*27 (cluster II) seemed to be associated with high susceptibility to gastrointestinal trichostrongyle nematodes, whereas the allele group Clgl DRB*19/*23/*26 (cluster II) and the allele Clgl DRB*21 (group III) are associated with high resistance to these nematodes. This suggests their functional difference in resistance to these prevalent nematode species. Thereby, taking the parasite data into account, one might argue that the allele group Clgl DRB*19/*23/*26 belonging to cluster II also seems to be associated with high resistance to trichostrongyle nematodes and thus still has an adaptive value in terms of parasite resistance. But, in fact, we observed a strong linkage between the Clgl DRB*26 allele out of this group and the allele Clgl DRB*21, so high resistance of these cluster II alleles might just be an artefact of close linkage. If this is assumed, then the cluster II alleles included in this parasite-specific analysis (Clgl DRB*20/*27 and Clgl DRB*24) would show higher parasite loads than group III alleles Clgl DRB*17 and Clgl DRB*21, which would be consistent with the overall picture on the evolutionary processes.
Putting all pieces of this puzzle together, our study may have revealed a snapshot in an evolutionary process of ongoing birth-and-death evolution. The allele cluster I might have lost its function and is already silenced, cluster II is loosing its adaptive value in terms of gastrointestinal nematode resistance, while a third group of alleles indicates all signs of classical functional MHC alleles. Ongoing studies will focus on expression divergence of Clgl DRB alleles in relation to parasite load to shed light in the ongoing birth-and-death process of MHC alleles in the bank vole.
References
Amills M, Jimenez N, Jordana J, Riccardi A, Fernandez-Arias A, Guiral J, Bouzat JL, Folch J, Sanchez A (2004) Low diversity in the major histocompatibility complex class II DRB1 gene of the Spanish ibex, Capra pyrenaica. Heredity 93:266–272
Apanius V, Penn D, Slev PR, Ruff LR, Potts WK (1997) The nature of selection on the major histocompatibility complex. Crit Rev Immunol 17:179–224
Baker CS, Vant MD, Dalebout ML, Lento GM, O’Brien SJ, Yuhki N (2006) Diversity and duplication of DQB and DRB-like genes of the MHC in baleen whales (suborder: Mysticetl). Immunogenetics 58:283–296
Barnard CJ, Kulis K, Behnke JM, Bajer A, Gromadzka-Ostowska J, Stachon M, Sinski E (2003) Local variation in helminth burdens of bank voles (Clethrionomys glareolus) from ecologically similar sites: temporal stability and relationships with hormone concentrations and social behaviour. J Helminthol 77:185–195
Behnke JM, Barnard CJ, Bajer A, Bray A, Dinmore J, Frake K, Osmond J, Race T, Sinski E (2001) Variation in the helminth community structure in bank voles (Clethrionomys glareolus) from three comparable localities in the Mazury Lake District region of Poland. Parasitology 123:401–414
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years. J Evol Biol 16:363–377
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39
Bryja A, Galan M, Charbonnel N, Cosson JF (2006) Duplication, balancing selection and trans-species evolution explain the high levels of polymorphism of the DQA MHC class II gene in voles (Arvicolinae). Immunogenetics 58:191–202
Bujalska G, Hansson L (2000) Bank vole biology: recent advances in the population biology of a model species. Pol J Ecol 48:1–235
Edwards SV, Chesnut K, Satta Y, Wakeland EK (1997) Ancestral polymorphism of Mhc class II genes in mice: implications for balancing selection and the mammalian molecular clock. Genetics 146:655–668
Ellegren H, Hartman G, Johansson M, Andersson L (1993) Major histocompatibility complex monomorphism and low levels of DNA fingerprinting variability in a reintroduced and rapidly expanding population of beavers. Proc Natl Acad Sci USA 90:8150–8153
Froeschke G, Sommer S (2005) MHC class II DRB variability and parasite load in the striped mouse (Rhabdomys pumilio) in the Southern Kalahari. Mol Biol Evol 22:1254–1259
Go Y, Satta Y, Kawamoto Y, Rakotoarisoa G, Randrianjafy A, Koyama N, Hirai H (2003) Frequent segmental sequence exchanges and rapid gene duplication characterize the MHC class I genes in lemurs. Immunogenetics 55:450–461
Gu X, Nei M (1999) Locus specificity of polymorphic alleles and evolution by a birth-and-death process in mammalian MHC genes. Mol Biol Evol 16:147–156
Harf R, Sommer S (2005) Association between major histocompatibility complex class II DRB alleles and parasite load in the hairy-footed gerbil, Gerbillurus paeba, in the Southern Kalahari. Mol Ecol 14:85–91
Haukisalmi V, Henttonen H (1993a) Coexistence in helminths of the bank vole Clethrionomys glareolus. I. Patterns of co-occurrence. J Anim Ecol 62:221–229
Haukisalmi V, Henttonen H (1993b) Coexistence in helminths of the bank vole Clethrionomys glareolus. II. Intestinal distribution and interspecific interactions. J Anim Ecol 62:230–238
Haukisalmi V, Henttonen H (2000) Variability of Helminth assemblages and populations in the bank vole Clethrionomys glareolus. Pol J Ecol 48:219–231
Hedrick PW (1999) Balancing selection and MHC. Genetica 104:207–214
Hedrick PW (2002) Pathogen resistance and genetic variation at MHC loci. Evolution 56:1902–1908
Horne TJ, Ylönen H (1996) Female bank voles (Clethrionomys glareolus) prefer dominant males; but what if there is no choice? Behav Ecol Sociobiol 38:401–405
Hörnfeldt B (2004) Long-term decline in numbers of cyclic voles in boreal Sweden: analysis and presentation of hypotheses. Oikos 107:376–392
Hughes AL (1999) Adaptive evolution of genes and genomes. Oxford University Press, New York
Hughes AL, Nei M (1988) Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335(86186):167–170
Hughes AL, Nei M (1989) Nucleotide substitution at major histocompatibility complex class II loci: Evidence for overdominant selection. Proceedings of the national Academy of Science, USA 86(3):958–962
Hughes AL, Friedman R (2004) Recent mammalian gene duplications: robust research for functionally divergent gene pairs. J Mol Evol 59:114–120
Jarvi SI, Tarr CL, Mcintosh CE, Atkinson CT, Fleischer RC (2004) Natural selection of the major histocompatibility complex (MHC) in Hawaiian honeycreepers (Drepanidinae). Mol Ecol 13:2157–2168
Jukes TH, Cantor CR (1969) evolution of protein molecules. In: Munroe HN (ed) Mammalian protein metabolism. Academic, New York
Klein J (1987) Origin of the major histocompatibility complex polymorphism: the trans-species hypothesis. Hum Immunol 19:155–162
Klein J, Sato A (1998) Birth of the major histocompatibility complex. Scand J Immunol 47:199–209
Klein J, Sato A, O’hUigin C (1998) Evolution by gene duplication in the major histocompatibility complex. Cytogenet Cell Genet 80:123–127
Klein J, Bontrop RE, Dawkins RL, Erlich HA, Gyllesten UB, Heise ER, Jones PP, Parham P, Wakelane EK, Watkins DI (1990) Nomenclature for the major histocompatibility complexes of different species, a proposal. Immunogenetics 31:217–219
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Li W-H, Yang J, Gu X (2005) Expression divergence between duplicated genes. Trends Genet 21:602–607
Martinsohn JT, Sousa AB, Guethlein LA, Howard JC (1999) The gene conversion hypothesis of MHC evolution: a review. Immunogenetics 50:168–200
Meyer-Lucht Y, Sommer S (2005) MHC diversity and the association to nematode parasitism in the yellow-necked mouse (Apodemus flavicollis). Mol Ecol 14:2233–2243
Michaux J, Reyes A, Catzeflis FM (2001) Evolutionary history of the most speciose mammals: molecular phylogeny of muroid rodents. Mol Biol Evol 18:2017–2031
Miller HC, Lambert DM (2004) Gene duplication and gene conversion in class II MHC genes of New Zealand robins (Petroicidae). Immunogenetics 56(3):178–191
Miska KB, Wright AM, Lundgren R, Sasaki-McClees R, Osterman A, Gale JM, Miller RD (2004) Analysis of a marsupial MHC region containing two recently duplicated class I loci. Mamm Genome 15:851–864
Musolf K, Meyer-Lucht Y, Sommer S (2004) Evolution of MHC-DRB class II polymorphism in the genus Apodemus and a comparism of DRB sequences within the family Muridae (Mammalia: Rodentia). Immunogenetics 56:420–426
Nei M (2005) Selectionism and neutralism in molecular evolution. Molecular Biology & Evolution 22(12):2318–2342
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152
Nei M, Gu X, Sitnikova T (1997) Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci USA 94:7799–7806
Norrdahl K, Korpimäki E (2002) Changes in population structure and reproduction during a 3-yr population cycle of voles. Oikos 96:331–345
Oliver MK, Piertney SB (2006) Isolation and characterization of a MHC class II DRB locus in the European water vole (Arvicola terrestris). Immunogenetics 58:390–395
Parham P, Otha T (1996) Population biology of antigen presentation by MHC class I molecules. Science 272:67–74
Phillips RB, Zimmerman A, Noakes MA, Palti Y, Morasch MR, Eiben L, Ristow SS, Thorgaard GH, Hansen JD (2003) Physical and genetic mapping of the rainbow trout major histocompatibility regions: Evidence for duplication of the class I region. Immunogenetics 55:561–569
Piertney SB, Oliver MK (2006) The evolutionary ecology of the major histocompatibility complex. Heredity 96:7–21
Reusch TB, Langfors A (2005) Inter-and intralocus recombination drive MHC class IIb gene diversification in a teleost, the three-spined stickleback Gasterosteus aculeatus. J Mol Evol 61:531–541
Reusch TBH, Schaschl H, Wegner KM (2004) Recent duplication and inter-locus gene conversionin major histocompatibility class II genes in a teleost, the three-spined stickleback. Immunogenetics 56:427–437
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Richman AD, Herrera LG, Nash D (2002) Characterization of Peromyscus MHC class II beta sequences by ligation-anchored RT-PCR and denaturing gradient gel electrophoresis. Eur J Immunogenet 29:213–217
Richman AD, Herrera LG, Nash D, Schierup MH (2003) Relative roles of mutation and recombination in generating allelic polymorphism at an MHC class II locus in Peromyscus maniculatus. Genet Res 82:89–99
Sachs I (1992) Angewandte Statistik, 7th edn. Springer, Berlin, Heidelberg New York
Sawyer SA (1999) GENECONV: A computer package for the statistical detection of gene conversion. Department of Mathematics, Washington University, St. Louis
Schad J, Sommer S, Ganzhorn JU (2004) MHC variability of a small lemur in the littoral forest fragments of Southeastern Madagascar. Conservation Genetics 5:299–309
Schad J, Ganzhorn JU, Sommer S (2005) Parasite burden and constitution of major histocompatibility complex in the Malagasy mouse lemur, Microcebus murinus. Evolution 59:439–450
Schaschl H, Suchentrunk F, Hammer S, Goodman SJ (2005) Recombination and the origin of sequence diversity in the DRB MHC class II locus in chamois (Rupicapra spp.). Immunogenetics 57:108–115
Schwensow N, Fietz J, Dausmann K, Sommer S (2007) Neutral versus adaptive genetic variation in parasite resistance: importance of MHC-supertypes in a free-ranging primate. Heredity (in press)
Seddon JM, Baverstock PR (2000) Evolutionary lineages of RT1.Ba in the Australian Rattus. Mol Biol Evol 17:768–772
Smulders MJM, Snoek LB, Booy G, Vosman B (2003) Complete loss of MHC genetic diversity in the common hamster (Cricetus cricetus) population in The Netherlands. Consequences for conservation strategies. Conservation Genetics 4:441–451
Sommer S (2003) Effects of habitat fragmentation and changes of dispersal behaviour after a recent population decline on the genetic variability of noncoding and coding DNA of monogamous Malagasy rodent. Mol Ecol 12:2845–2851
Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2:1–18
Stear MJ, Innocent GT, Buitkamp J (2005) The evolution and maintenance of polymorphism in the major histocompatibility complex. Vet Immunol Immunopathol 108:53–57
Ylönen H, Koskela E, Mappes T (1997) Infanticide in the bank vole (Clethrionomys glareolus): occurrence and the effect of familiarity on female infanticide. Ann Zool Fenn 34:259–266
Zhang J (2003) Evolution by gene duplication: an update. Trends Ecol Evol 18:292–298
Acknowledgements
We thank ‘Landesamt für Natur- und Umwelt-Schleswig Holstein’ and the Kreisrevierförster J. Stäcker for permission to carry out this project. An anonymous referee provided helpful comments on a former version of this manuscript. We are grateful to I. Tomaschweski, Y. Meyer-Lucht, J.U. Ganzhorn and all field assistants for supporting our project.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Axtner, J., Sommer, S. Gene duplication, allelic diversity, selection processes and adaptive value of MHC class II DRB genes of the bank vole, Clethrionomys glareolus . Immunogenetics 59, 417–426 (2007). https://doi.org/10.1007/s00251-007-0205-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-007-0205-y