
Abstract
It has been known for decades that proteins undergo conformational changes in response to binding ligands. Such changes are usually accompanied by a loss of entropy by the protein, and thus conformational changes are integral to the thermodynamics of ligand association. Methods to detect these alterations are numerous; here, we focus on the sedimentation velocity (SV) mode of AUC, which has several advantages, including ease of use and rigorous data-selection criteria. In SV, it is assumed that conformational changes manifest primarily as differences in the sedimentation coefficient (the s-value). Two methods of determining s-value differences were assessed. The first method used the widely adopted c(s) distribution to gather statistics on the s-value differences to determine whether the observed changes were reliable. In the second method, a decades-old technique called “difference SV” was revived and updated to address its viability in this era of modern instrumentation. Both methods worked well to determine the extent of conformational changes to three model systems. Both simulations and experiments were used to explore the strengths and limitations of the methods. Finally, software incorporating these methodologies was produced.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ligand-induced conformational changes in proteins have been documented in many instances. ABC transporters (Davidson and Maloney 2007), gated ion channels (Catterall et al. 2017), G-protein-coupled receptors (Deupi and Standfuss 2011), and enzymes (Gerhart and Schachman 1968; Bennett and Steitz 1978) are just a few examples of proteins that undergo structural rearrangements as intrinsic parts of their respective functional cycles. Additionally, there has been recent interest in targeting intrinsically disordered proteins with small molecules that induce conformational shifts (e.g., (Krishnan et al. 2014)). These structural transformations have a number of purposes. For example, in enzymes, they may trigger allosteric communication between an effector binding site and an active site (Kamata et al. 2004). Some enzymes might use conformational changes to shelter substrates or products from the bulk solvent, preventing side reactions with water (Dwyer and Hellinga 2004; Fawaz et al. 2011; Khan et al. 2017). Small-molecule-binding proteins, like the bilobed periplasmic ligand-binding proteins (LBPs), feature significant interlobe motions as an integral part of the ligand-binding mechanism; these changes serve to maximize protein–ligand contacts, desolvate the ligand, and provide a physical barrier to dissociation (Mao et al. 1982; Felder et al. 1999; Dwyer and Hellinga 2004). Generally, unliganded proteins that undergo conformational changes exist in an ensemble of structural states which collectively may be called the “open” state. Once a ligand binds, this variation collapses into just one or a few states, which can be characterized as “closed” [notably, some binding events lead to a less compact conformation (Harris and Winzor 1988)]. There is an obvious loss of entropy in transitioning from the open to the closed state(s), and any thorough thermodynamic study of the ligand-binding process must take this into account.
The methods to study protein conformational changes are manifold. Obviously, X-ray crystallography of the unbound and bound states can and has reported on the conformational changes (e.g., Bennett and Steitz 1978; Kamata et al. 2004), with the omnipresent caveat that the crystal lattice may influence conformations. Recent technological and analytic advances in cryo-electron microscopy (Bai et al. 2013; Li et al. 2013) have allowed this method to monitor ligand-induced conformational changes both gross and subtle (e.g., Gutmann et al. 2018; Shang et al. 2019; Uchikawa et al. 2019), but the method involves expensive, specialized equipment and substantial technical expertise at present. Multidimensional solution NMR spectroscopy offers insights into ligand-induced changes, but it can involve sometimes expensive labeling techniques for the proteins (Persons et al. 2018). Another solution method is small-angle X-ray scattering (SAXS). The calculation of the radius of gyration (Rg) from the Guinier region of a scattering profile is an excellent means to monitor conformational changes (Newcomer et al. 1981; Borrok et al. 2009), but the need for specialized equipment (usually a synchrotron) and the possibility of radiation damage limit the applicability of SAXS. Finally, several modern methods, such as second-harmonic generation (SHG) (Moree et al. 2015), and surface-acoustic wave (SAW) (Länge et al. 2008), and double electron–electron resonance (DEER) (Jeschke 2012), require immobilization on a surface and/or labeling the protein (with a proprietary dye or a spin label). Such modifications to the protein’s environment may be suited to some proteins, but not all.
It was recognized decades ago that the sedimentation velocity (SV) mode of analytical ultracentrifugation could be used to monitor protein conformational changes (Richards and Schachman 1959; Kirschner and Schachman 1971b) in solution with no perturbations to the macromolecule. This is because the migration velocity of proteins in a centrifugal field (monitored without labeling and quantified by the sedimentation coefficient, s) is governed in part by their respective hydrodynamic radii (RH). These latter quantities will vary commensurately with the proteins’ conformational changes. That is, when compared side-by-side, if the same protein has different s-values in the presence and absence of ligand, it is likely due to a conformation difference (after mass changes are accounted for; if ligand-induced oligomerization is present, this analysis is not applicable). Historically, two possibilities to quantify these changes were considered. In the first, two independent experiments were carried out, plus and minus ligand (e.g., see Gerhart and Schachman 1968; Oberfelder et al. 1984; Jacobsen and Winzor 1997). The s-values were determined, and changes were attributed to conformational differences. However, this was deemed too imprecise to quantify small conformational changes, motivating the second method. The advent of interferometric monitoring of the protein-concentration profiles led Schachman and colleagues to propose a technique called “difference sedimentation velocity” (DSV) (Richards and Schachman 1959; Kirschner and Schachman 1971b). In this method, protein is placed in both sectors of a dual-sectored centerpiece (Fig. 1a). In one sector, a ligand hypothesized to induce a conformational change upon binding is added; in the other, a similarly sized, but non-binding ligand is included. Using the interference optics, monochromatic light is passed simultaneously through both sectors, and these slits of light are recombined to form a radial interference pattern. In this way, the radially dependent refractive-index differences between the sectors can be measured. If there is no conformational change, then the macromolecular solutes in both sectors migrate identically, and no difference is recorded. However, a ligand-induced conformational change can lead to a different (usually faster) velocity in the ligand-containing sector only, resulting in a Gaussian-like difference pattern (Fig. 1b). This method was deemed sensitive enough to detect very small Δs-values (on the order of 0.5%). DSV was successfully used to measure conformational changes in aspartyl transcarbamylase (Kirschner and Schachman 1971a), ribonucleotide reductase (Singh et al. 1977), and several other proteins (Kirschner and Schachman 1971b).

Difference sedimentation velocity. a Signal profiles of the individual sectors. A schematic of the standard sedimentation velocity centerpiece is shown inset, with the reference sector colored red and the sample sector blue. A vector of centrifugal force is shown as a white arrow. Simulated SV data are shown. A 9.5% difference between the sedimentation coefficients in the reference (4.2 S) and sample (4.6 S) sectors was simulated. Profiles originating from the reference and sample sectors are respectively colored according to the inset centerpiece diagram and inset legend. Both simulations originated from the same meniscus value (6.1 cm) and both are shown at the same time point after the commencement of centrifugation (7090 s). b The difference curve that results from subtracting the “reference line” from the “sample” line in part a. The curve does not return to zero on the right-hand side; this depicts the difference in radial dilution between the sectors. If there were no difference in the sedimentation coefficients, this offset would be absent
Both methods described above were introduced in an era of AUC in which graphical methods were used to analyze data. For both, data from the Beckman Model-E centrifuge were recorded on photographic plates. Measurements were taken from these plates to calculate the values needed for the respective analysis; for SV, the maxima of differential curves obtained using Schlieren optics, and for DSV, the first moment of the interferometric difference concentration distribution. Often, technology such as a microcomparator was used to digitize the measurements obtained from the photographs. Thus, few measurements could be taken per run and analysis could be arduous.
However, modern methods of data acquisition and analysis have significantly improved. Data are acquired quickly and digitally, enabling the collection of hundreds of scans per AUC experiment. No external, manual measurement of the data is necessary. Also, the introduction of modern computerized data analysis, particularly the c(s) method (Schuck 2000), has allowed rapid assessment of s-values by fitting sedimentation models directly to the digitized data. Despite these advances, the use of AUC to detect small conformational changes in proteins is uncommon. Some notable exceptions include iron-regulatory proteins (Yikilmaz et al. 2005), matrilin-3 (Fresquet et al. 2007), canine plasminogen (Kornblatt and Schuck 2005) and 5-enolpyruvylshikimate-3-phosphate synthase (Borges et al. 2006).
Our interest in these conformational changes was spawned by recently determined X-ray crystal structures featuring substantial ligand-induced rearrangements. Structures of a mutated glucose-binding protein (the product of gene tp0684; called “TpMglB-2WA” herein) from the syphilis spirochete, Treponema pallidum, suggested that the protein undergoes a domain closure featuring a rotation of approximately 39° upon binding d-glucose (Brautigam et al. 2018), in accord with the known properties of this family of ligand-binding proteins from ABC transporters (Mao et al. 1982; Borrok et al. 2007).
In this report, we used modern AUC and computational methods to examine whether they could detect the conformational changes described above. Both the SV and DSV methods were applied to a model protein, bovine serum albumin, as well as to TpMglB-2WA. In all cases, we were able to detect small differences in sedimentation coefficients on the order of 2%. Our studies revealed a set of best practices and computational methods, some of which are encoded into a new, freely available software program called DiSECT.
Results
Hydrodynamic modeling
Throughout this work, we describe a scenario that could become common under the current state of the technologies surrounding structural biology. That is, that X-ray crystal structures of a protein in a liganded (or “holo”) and unliganded apo form are available, and they show a conformational change in the protein upon ligand binding. A natural question arises from such structures: does an alteration of similar magnitude occur in solution? We thus address how a combination of hydrodynamic modeling and SV experiments could be used to answer this question.
The availability of the apo- and holo- X-ray crystal structures of TpMglB-2WA (Brautigam et al. 2018) presented a good opportunity to address this question (Fig. 2). In the structures, the protein has an “open” appearance without ligand (Fig. 2a), but “closes” upon binding d-glucose (Fig. 2b). First, we chose to perform hydrodynamic modeling (de la Torre et al. 2000; Fleming and Fleming 2018) on the respective coordinate sets. These calculations would reveal the expected s-values for the unliganded and liganded versions of the protein. As pointed out by Errington and Rowe (2003), the s-values obtained by modeling efforts may not reflect the veracity of a given conformational state. For example, if the modeling predicted 3.31 S for the holo structure but the actual experiment on the holo protein showed a value of 3.45 S, this result does not necessarily confirm that the solution and crystal conformation are different. However, the same authors point out that the hypothetical magnitude of the change, what we termed the “hydrodynamically modeled Δs” (ΔsModel), predicted by modeling both forms observed in the crystal structures should prove reliable in predicting the solution behavior of the respective forms. Our focus, therefore, is in determining the expected Δs elicited by the “structural” ligand-induced conformational change, then comparing to the “solution” result later.

Crystal structures of TpMglB-2WA show a d-glucose-induced conformational change. Ribbons-style representations of the crystal structures are shown, with α-helices in red, β-strands as blue arrows, and regions without regular secondary structure in light blue. a Apo TpMglB-2WA. b TpMglB-2WA bound to d-glucose. The glucose molecule is depicted as spheres, with carbon atoms in black and oxygen in red
We used two methods to calculate sedimentation coefficients from the structural models. The first was the bead modeling encoded in the software HYDROPRO (de la Torre et al. 2000). In this program, care was taken to achieve the most accurate s-values from the modeling. For example, the mass of d-glucose was included in the holo-TpMglB-2WA model. As a result, the modeled s-value of apo-TpMglB-2WA was 3.23 S, while that of the holo form was 3.32 S. We also used the same structural models to calculate the respective s-values using a recently introduced convex-hull method (Fleming and Fleming 2018), which resulted in 3.39 S and 3.48 S for these two structural forms, respectively. Although it is interesting that the two calculations resulted in ~ 5% differences for the predicted s-values, the most important aspects of these results are that they both predicted an increase of the s-value in the presence of d-glucose and that they both predicted the value of ΔsModel would be 0.09 S. If we assume that each hydrodynamic simulation has an error of 1% and that the errors add in quadrature, the standard error in the ΔsModel would be 0.05 S.
Conformational changes: the SV method
SV general considerations
In the SV method, one aims to determine s-values from experiments conducted both with and without ligand present. After that is achieved, the difference should be calculated, with appropriate experimental and analytic errors taken into account. Obviously, in employing this method, researchers must aim for the most precise s measurements possible; but how is that achieved? Errington and Rowe (2003) enumerated the factors that could lead to imprecision and inaccuracy in s-values; they are not recapitulated here, but most of these factors disappear when comparative sedimentation experiments are performed simultaneously, i.e., side-by-side in the same instrument. Thus, because all comparative experiments demand high precision, we conducted them simultaneously in a single 8-hole AUC rotor. However, some factors affecting precision still remained. SV experiments are conducted in AUC “cells”, which consist of a dual-sectored centerpiece positioned between transparent windows. These cells must be inserted into the rotor and aligned precisely with respect to the vector of centrifugal force (Fig. 1a, inset). Thus, cell-to-cell shape inconsistencies and individual cell-alignment procedures can cause variability in the determination of s. Because these factors are specific to cells, determining reliable s-values for Δs determinations necessitates obtaining the averages of several measurements from different cells. Given the limitation of eight cells per experiment, the number of replicates can conveniently range between three and four (e.g., three cells containing ligand-free protein, and three containing ligand-bound protein). Because each individual s-value determination will have an accompanying analytical error, we prefer to calculate mean of these replicates using a scheme that weights each measurement with its respective analytic error (see “Methods”). In this paper, we will term the weighted mean of three or four s measurements “sav”, and the estimate of the weighted standard deviation from this calculation is “σav”.
SV model system
A traditional means to test whether a Δs can be reliably measured is to use a model system. In the past, the large (132 S) bushy stunt virus (BSV) has been used for this purpose (Kirschner and Schachman 1971b). To induce a small Δs consistent with a conformational change, D2O was introduced into only one of the samples to be compared, altering the mass of the virus along with the viscosity and density of the solution (Kirschner and Schachman 1971b). In this work, we employ the same strategy using bovine serum albumin (BSA), a much smaller macromolecule having an experimental s-value of about 4.3 S under dilute conditions.
Experimentally, we conducted eight SV experiments in a single run of the AUC. Four of the cells had BSA at 1 mg/mL in PBS buffer. Also, four of the cells contained BSA at an identical concentration, but with the PBS supplemented 4% (v/v) in D2O. Taking the changes in BSA mass (through deuterium exchange), solution density, and solution viscosity into account (Kirschner and Schachman 1971b), we expected a Δsav of approximately 0.08 S. To maximize the number of data points reporting on s, we used the Rayleigh interference optics exclusively and collected one concentration profile per minute. The absorbance optics were not employed, because their lower data density and longer time of acquisition (ca. 90 s per scan in our centrifuge) would have a significantly reduced the amount of data available for analysis. The data were analyzed by the c(s) methodology (Schuck 2000), and the individual s-values were obtained by integrating the respective c(s) distributions. The distributions showed that the protein was almost entirely monomeric and monodisperse (Fig. S1), and this latter observation was confirmed using a Bayesian analysis (Brown et al. 2007) (i.e., no microheterogeneity was observed; not shown). Analytic errors were assessed using a Monte Carlo procedure (see “Methods”) (Schuck 2016).
The results, summarized in Table 1, demonstrated that a difference in sav-values was observed to be 0.08 S, as expected. However, given the measurement and analysis errors, could this difference be the result of chance experimental variations? To explore this possibility, we used the results in conjunction with t-statistics to calculate that the probability of this difference occurring by chance is extraordinarily low (p = 2 × 10–9; two-sided Student’s t test). The SV method thus appears to be a reliable way to obtain a Δsav of this magnitude.
SV of TpMglB-2WA
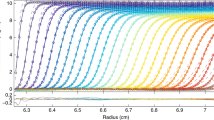
To measure sav for TpMglB-2WA in both the holo and apo forms, we conducted six SV experiments in a single run of the AUC. Three of the centrifugation cells contained 1.0 mg/mL TpMglB-2WA with 1 mM d-glucose, and the other three held the same concentration of protein, but also a ligand with no detectable affinity for TpMglB-2 (Brautigam et al. 2016), d-ribose, at 1 mM concentration. The same analytic workflow as used for BSA was employed here. These distributions showed that the preparations of TpMglB-2WA were essentially monodisperse; ~ 92% of the observed signal was from the monomeric form of this protein (Fig. S2). Visually, the d-glucose-containing samples always had a discernibly larger s-value (Fig. 3). The best s-values of TpMglB-2WA determined by integration of the c(s) distributions are shown in Table 2. They display consistently larger values for the d-glucose-containing samples, which comports with the ligand-induced closure of the cleft (Fig. 2) observed in crystal structures (Brautigam et al. 2018). We found that Δsav was 0.082 S (Table 2), which is not far from the ΔsModel calculated above (0.09 S). The probability that the measured difference of 0.082 S could exist by chance is very low (p = 0.0004 using a two-sided Student’s t test). The Δs that would be expected if the change were due the mass of the binding ligand alone can be calculated (Kirschner and Schachman 1971b); for the current case, that value is 0.02 S. The probability that the change was less than 0.02 S leading to the observed values being observed by chance was similarly low (p = 0.001, one-sided Student’s t test). Finally, the question can be asked about the consistency of the SV result with the ΔsModel. With the assumptions that the σav’s add in quadrature and that the σ of the modeled Δs is 0.05 S, we estimate that the probability of the 0.008 S difference between the ΔsModel and Δsav being chance is 0.45, i.e., there does not seem to be a reliable difference between the two values. The results thus seem consistent with the notion that a conformational change of similar magnitude to that observed in the TpMglB-2WA structures is also observed in solution.

Two c(s) distributions for TpMglB-2WA. The experiments were conducted in the presence of genuine ligand (d-glucose) or a mock ligand (d-ribose) as indicated in the inset legend
Conformational changes: the difference sedimentation velocity method
Overall considerations and strategy
In the late 1950s, Schachman and coworkers demonstrated that the Rayleigh interference optical system was uniquely capable of detecting very small changes in s-values (Richards and Schachman 1959). The strategy was simple: introduce identical concentrations of the subject protein into the reference and sample sectors of a standard centerpiece, with the only difference between the two being the presence of a conformation-changing ligand in one sector and not in the other (Richards and Schachman 1959; Kirschner and Schachman 1971b). The advantage of this approach is that it determines the refractive-index difference between the reference sector and the sample sector of a standard centerpiece. If there is a difference in the sedimentation velocity of the solutes in the sectors (Fig. 1a), it can be identified as a time-dependent increase in the first moment of the resulting difference curve (Fig. 1b) (Richards and Schachman 1959; Kirschner and Schachman 1971b). Hence, these moments can be calculated and plotted as a function of time (radial migration), with any slope indicating a difference in s of the material in the sectors (Fig. 4). This method, therefore, measures Δs directly, without the need of calculating individual s-values and subtracting one from another, as was done above. Although refinements on the procedure were made throughout the 1970s (Kirschner and Schachman 1971a, b; Skerrett 1975; Rees et al. 1977), they were always accomplished with older hardware (i.e., the Beckman Model-E analytical ultracentrifuge). Because of the laboriousness of the data acquisition and analysis, this type of analysis was usually performed using 5–10 scans.

Linearized first-moment data for BSA and TpMglB-2WA. In both cases, the circles represent the data, and black lines are fits to those data. Blue circles are for the actual experiment, and red ones are for the negative control experiment. a BSA data. Data are extrapolated to x values of 0.0; the y-intercept should depict the difference in meniscus position between the reference and sample sectors (Richards and Schachman 1959; Kirschner and Schachman 1971b). The slopes are the respective Δs/s̄ values, which have been transformed to Δslin in the text. The negative slope for the “actual” data (blue circles) is a consequence of the reference sector being filled slightly more than the sample sector (this was inadvertent). b The TpMglB-2WA data
Previously (Brautigam et al. 2016), we employed this linearized DSV method to determine whether wild-type TpMglB-2 underwent a conformational change when bound to d-glucose. In this work, we sought to refine the method for the modern hardware, examine critical control experiments, and explore the possibility of directly analyzing the DSV curves with differenced Lamm-equation solutions in addition to integrating them and performing the linear-regression analysis employed decades ago. Critically, the modern hardware allows us to perform the analyses on tens to hundreds of scans, an advantage that should improve accuracy and precision.
Experimentally, two simultaneously performed studies are essential to provide enough information for the analysis. First, as is standard DSV practice, one centerpiece sector should be filled with unliganded protein, and the second with the identical concentration of protein along with the ligand that induces the conformational change. It is useful to include in the “unliganded” sample a small molecule that has a similar molar mass to the ligand, but does not bind to the subject protein; this expedient helps to prevent the detection of refractive-index differences due to presence of a small molecule in only one sector (Oberfelder et al. 1985). The second experiment is a control: using exactly the same concentration as present in the DSV experiment, a standard AUC experiment (i.e., buffer in the reference sector, unliganded protein in the sample sector) should be carried out and analyzed to establish the magnitude of the signal (ΔJU) and the s-value (sU) of the unliganded solute under the experimental conditions. We called this control the “SAM Control” (for “s And Magnitude”). It is necessary to input these values and fix the magnitude in the analysis method described below. Another useful, but not essential, control experiment is provided by applying identical solutions to both sectors. Thus, there should be no Δs between the sectors, and analyses of these data serve as a test of the user’s technique and apparatus.
The analysis of the DSV data acquired as described above (or simulated) was carried out using an automated algorithm implemented as a stand-alone Python program (“DiSECT”, see below). Upon starting the program, the information from the SAM control was inputted, and then the data were loaded followed by choosing the menisci and the radial fitting limits. After that, the algorithm was actuated. It automatically determined the data range that was useful for the analysis from hundreds of available scans. It calculated time-invariant noise (Schuck and Demeler 1999) in the data and removed it. The normalized first moments of the baseline-subtracted difference curves were calculated and tabulated, and a linear regression was performed on these (Fig. 4), with the quantity Δs/\(\overline{s}\) (where \(\overline{s}\) is the mean s-value of the material in the reference and sample sectors) being derived from the slope of the regressed line (Kirschner and Schachman 1971b). The values obtained from this linearized analysis were used as the starting point for a direct analysis of the difference-curve data (Fig. 5) using the transport terms of an approximate analytic solution of the Lamm equation (Behlke and Ristau 2002). The analysis can be performed in just a few seconds with hundreds of scans and minimal user input.

DSV results. In both parts, the upper graph shows the noise- and baseline-subtracted data (circles) and fits to those data (black lines). For clarity, only every 5th analyzed scan is shown. Colors represent the respective time of the scans, from early (purple) to late (red). The bottom graph shows the residuals between the data and the fits as respectively colored circles. a The BSA D2O/H2O experiment. b The TpMglB-2WA d-ribose /d-glucose experiment. The differing “sign” of the DSV curves is a result of a slight (and inadvertent) overfilling of the sample sector in the case of BSA and deliberate underfilling of the sample sector for TpMglB-2WA experiment
DSV of simulated data
As a first test of this methodology, we simulated noisy, BSA-like data using an independent data-generation algorithm. We chose the numerical Lamm-equation simulations available in SEDFIT (Brown and Schuck 2008). Because that program does not simulate difference curves like that in Fig. 1b directly, a custom simulation workflow was established (see “Methods”). The simulation parameters are given in Table 3. Note that the reference and sample sectors were given different menisci; a deliberate meniscus mismatch is commonly introduced into DSV experiments to give the difference curves a significant magnitude, facilitating the analysis (Kirschner and Schachman 1971b; Oberfelder et al. 1985). No SAM experiment was simulated, as sU and ΔJU were known to the user.
We first simulated a system in which the ligand was placed in the sample sector only, causing a change of roughly 1.8% in the sedimentation coefficient (0.08 S). To test the stability of the algorithm for all likely configurations, four scenarios were tested: the four combinations possible from the reference/sample menisci mismatches and ligand-induced protein expansion vs. compaction (scenarios 1–4 in Table 3). In comparing the values of Δs obtained from the linearized fit (Δslin) and that obtained from the direct fit to the difference data (ΔsDSV), we found that the latter methodology consistently lent more accuracy and precision to the analysis. All of the ΔsDSV values obtained were within 1.25% of the simulated value, suggesting that the analytic strategy is capable of arriving at robust estimates of Δs using noisy but otherwise ideal data.
We examined a fifth scenario under which the sedimentation coefficients were identical in both sectors. Both the linearized and direct-fitting approaches correctly identified that lack of a sedimentation-coefficient difference between the sectors (Table 3). Thus, it appears that these methods do not easily yield false-positive results.
DSV on BSA
In analogy to the preliminary experiments above that explored differences in sedimentation coefficient induced by the addition of D2O to a solution of PBS, we conducted DSV experiments in a similar mode. That is, in this DSV experiment, a solution of 4.5 mg/mL of BSA in PBS was placed into the sample sector of the centerpiece, while an identical concentration of BSA was introduced into the reference sector, but this solution was 4% (v/v) D2O. A SAM experiment was also conducted along with a negative control with BSA and 4% D2O in both sectors.
The analysis of the SAM experiment demonstrated that 15.032 fringes of material were present, and the best s-value for the BSA monomer was 3.974 S. The lower s-value for the BSA monomer compared to the SV experiment described above can be ascribed to non-ideality in the more concentrated BSA solutions used in this part. After that, the remainder of the analysis was conducted using the software that contains the analytic methods described herein, entitled DiSECT. The analytic workflow was: (1) Start DiSECT. (2) Load the DSV data. (3) Input sU and ΔJU. (4) Adjust the positions of the menisci and fitting limits on the data. (5) Actuate the execution of the algorithm. And (6) refine and finalize the results. The results (Figs. 4a, 5a) showed that the algorithm performed well (Table 4). For the negative control experiment, ΔsDSV was − 0.0069 [− 0.0080, − 0.0058] S (throughout this work, 95% confidence intervals obtained by an automated error-surface projection method are presented in square brackets), showing a slightly negative bias from the expected value of 0.0 S. For the comparison between D2O and H2O, ΔsDSV was 0.0701 [0.0689, 0.0712] S. Again, this was slightly lower than the expected value of 0.08 S. Instead of just a few scans, we were able to accomplish these analyses with 167 and 134 scans for the control and heavy water-comparison experiments, respectively.
DSV of TpMglB-2WA
We performed three simultaneous AUC experiments with TpMglB-2WA at 4.4 mg/mL, chosen to be analogous to the BSA studies presented above. The first experiment compared the sedimentation of the protein in the presence of d-ribose vs. d-glucose (the sugars were included at a concentration of 1 mM). Protein with d-ribose was introduced into the reference sector, and protein with d-glucose was in the sample sector. Another experiment was the negative control: d-glucose was included with the protein in both sectors of the centerpiece, and thus no Δs should be detectable. Finally, the SAM experiment featured reference buffer in the reference sector and unliganded 4.4 mg/mL TpMglB-2WA in the sample sector. The interference optics were used, and one concentration profile was acquired per minute.
With the data in hand, we followed the same analytic procedure that had been established with the BSA experiments (Figs. 4b, 5b; Table 4), using 88 scans for the negative control and 95 scans for the d-ribose/d-glucose experiment. The values of sU and ΔJU were 3.135 S and 14.041 fringes, respectively. The Δslin was 0.0645 [0.0619, 0.0680] S and the ΔsDSV was 0.067 [0.064, 0.071] S (Figs. 4b, 5b). Thus, there was clear evidence of a difference in the sedimentation coefficient between the d-glucose-bound form of TpMglB-2WA and the same protein in the presence of d-ribose.
The question remains, however, whether the observed difference accords with ΔsModel (the hydrodynamically modeled Δs) as calculated above. The ΔsDSV is well above the value calculated if d-glucose bound with no accompanying conformational change (0.02 S, vide supra), and thus the conformational change was reliably detected by DSV. With significant assumptions, a two-sided t test may be performed to examine the possibility of a real difference between ΔsModel (0.09 ± 0.05 S) and ΔsDSV (0.067 ± 0.002 S, with the σDSV estimated as the upper limit of the 95% confidence interval minus the best refined value divided by two). With these assumptions in place, p = 0.36. Statistically, therefore, it appears that the ΔsDSV is consistent with the expected conformational change (or, more precisely, it cannot be stated confidently that there is any difference between ΔsModel and ΔsDSV).
Discussion
In this study, we have used two means to detect sedimentation coefficient differences between the same protein in two different solutions, with the ultimate goal of providing a modern update to the classic literature on monitoring ligand-induced macromolecular conformational changes using SV. The first method was rooted in the c(s) analysis (Figs. 3, S1, and S2). In examining our results, we can recommend some best practices to maximize the precision of the measured s-values and thus the user’s confidence in the veracity of the posited change in conformation. At least three replicates each of mock-liganded and ligand-bound proteins should be examined in a single AUC experiment (i.e., a total of 6–8 centrifugation cells should be used). It is critical that all experiments be done at identical protein concentration, as concentration differences can lead to apparent s-value changes. Doing all at the same time eliminates many sources of experimental variability (Errington and Rowe 2003), and the recommended number of replicates allows the user to achieve meaningful statistics that can account for cell-to-cell variability. The interference optics should be used exclusively for data acquisition when possible, because the speed of data acquisition allows more data to be collected, which improves the precision in s. Finally, we suggest a Monte Carlo-based protocol (Schuck 2016) to determine the confidence interval on each measured s-value, followed by the determination of a weighted mean to obtain the best estimate of s from each set of replicates, sav. Standard statistics on Δsav can yield the confidence with which the conformational change may be stated. In our case, Δsav was detected very reliably for both the BSA test and TpMglB-2WA.
We also employed a new DSV method to detect changes in conformation (Figs. 4, 5), manifested in the quantity ΔsDSV. The method relies on fitting the Gaussian-like DSV data directly, rather than the linearized analysis first employed by Schachman and colleagues (Richards and Schachman 1959; Kirschner and Schachman 1971b). Sedimentation-coefficient differences were reliably detected in these experiments. Here, we found that replicates are not obligatory. This is because the method directly detects the difference in sedimentation velocity of two solutes side-by-side in a single centerpiece. With careful experimentation, incorrectly detecting a significant ΔsDSV is very unlikely. The experiment, as we envision it, usually employs only three centrifugation cells: one “SAM” experiment, containing buffer in the reference sector and the macromolecule under study in the sample sector, which allows the s-value and signal magnitude of the sample to be elucidated; one experimental cell, having macromolecule plus a mock ligand in the reference sector and the macromolecule plus the true ligand in the sample sector; and one negative control to check for systematic offsets in ΔsDSV, containing macromolecule with the true ligand in both sectors. As before, the macromolecular concentrations in all samples must be identical. Following the suggestions of earlier practitioners (Kirschner and Schachman 1971b; Oberfelder et al. 1985), we deliberately introduced a meniscus mismatch to the latter two experiments, allowing easier integration and interpretation of the resulting difference curves. In the current instance, we detected a ΔsDSV in TpMglB-2WA upon ligand binding that was commensurate with our hydrodynamic calculations.
Importantly, we have undertaken our DSV experiments with no special equipment, adjustments, or customized apparatus. The goal was to ascertain whether the method could be implemented using the currently deployed centrifuge by a careful experimenter who is not an expert in interference optics. The success of the experiment demonstrates that conformational changes of the magnitude studied herein are readily detected in DSV experiments without extraordinary measures.
Our DSV analytic protocol includes performing a “linearized” analysis (Kirschner and Schachman 1971a) to obtain estimates for Δs, then fitting the interferometric data directly. This latter part was accomplished using the transport terms of an equation formulated by Behlke and Ristau (2002), which has also been used in other contexts [e.g., in the SVEDBERG program by Philo (1997)]. For each scan analyzed, our algorithm calculates two concentration profiles and subtracts them, allowing the fitting of these difference curves to the DSV data. We found that most parameters in the fitting were well-specified by the data, i.e., the refinements of those values did not lead to unreliable results. The overall concentration, ΔJU, was not one of these parameters. In our tests of the algorithm, it was strongly correlated with other parameters, and thus it is imperative to find its value via the SAM experiment and fix it during the analysis.
Despite the positive DSV result, there is clearly room for improvement of the DSV protocol. First, as implemented above, the method consumes nearly 10 mg of protein (~ 2 mL at ~ 5 mg/mL using our three-experiment protocol). In preliminary studies (not shown), we found that concentrations of protein down to 2 mg/mL could reliably report on ΔsDSV. Also, changes in centerpiece design featuring narrower sectors (Desai et al. 2016; To et al. 2019) are envisioned, thus lowering the volume (and hence total mass of material) necessary to conduct a DSV experiment.
But the most serious drawback that we observed in our DSV studies was a systematically negative bias was in the observed ΔsDSV values (Table 4). For example, the apparent error in the measurement of ΔsDSV for TpMglB-2WA was − 18% (cf. Δsav and ΔsDSV, Tables 2, 4). The source of this bias is unknown at present. At least one candidate source errors of this magnitude can be immediately eliminated: the direct-fitting algorithm. Our simulations of DSV experiments (Table 3) demonstrated that the analytic method works well, and can only account for errors on the order of 1%. One possibility is in data preparation for the direct fitting, specifically with regard to the baseline subtraction. The TI noise elements that are part of the baseline are calculated from the last 10 scans of the experiment, but those can be well separated in time from the subset of scans that is analyzed in our method. Also, contaminating species existing within the diffusional envelope of the main species could alter the baseline calculation and the shape of the DSV data curve. Another potential source of the problem may lie in the modern interference optical system and other aspects of interferometric data collection. The alignment of the Rayleigh interference system and the alignment of the cells within the rotor was accomplished more crudely compared to the exacting methods employed by Schachman and colleagues (Richards and Schachman 1959; Kirschner and Schachman 1971b). The originators of the DSV technique outlined some other putative sources of data-collection errors, including light path-length differences in the assembled AUC cells and fringe “necking” or “bowing” (owing to large refractive-index gradients) (Kirschner and Schachman 1971b). We envision that systematic centerpiece flaws or fringe gradients induced by the deliberate meniscus mismatch could also contribute to the bias. A final possible source of error is differential non-ideality effects that manifest at the high concentrations used for the DSV studies. This could be systematically explored by examining the experimental sedimentation coefficient of TpMglB-2WA as a function of concentration in the presence of d-ribose and d-glucose to see if there are differing non-ideality constants. Means to improve the performance of the DSV experiments and calculations are currently under study.
Despite the slight bias in the DSV results, we found that both modern, AUC-based methods of detecting a ligand-induced conformational change explored in this study can yield acceptable results. The user may, therefore, choose the method to best meet experimental needs. The c(s)-based technique works well for systems in which sample quantity is limited or there is some noninteracting contaminant present (the distribution may be integrated to exclude the contaminant). However, this method can be laborious, and it requires the user to have at least six centrifugation cells and an eight-hole rotor on hand. Notably, the eight-hole rotor cannot achieve rotor speeds higher than 50,000 rpm, and thus small proteins and peptides may not be amenable to this approach. The DSV method requires only three cells and thus a four- or eight-hole rotor may be employed (enabling speeds up to 60,000 rpm). Using our approach, the latter data require only minimal processing, and the actual DSV analysis takes only seconds. The downside is the high (2–5 mg/mL) concentration and high purity (> 95%) of material required.
Two types of ligand-induced conformational changes in proteins are possible. In the first, the protein expands upon ligand binding, as in rabbit pyruvate kinase (Harris and Winzor 1988), manifesting as a smaller sedimentation coefficient in the presence of the ligand. This phenomenon is clear-cut evidence for a conformational change. In the second scenario, the protein contracts, causing its sedimentation coefficient to rise slightly; a classic example is acetyl transcarbamylase (Kirschner and Schachman 1971a), and it was also the case for TpMglB-2WA (Fig. 2) (Brautigam et al. 2018). However, another possible explanation is that ligand binding induces oligomerization of the protein; that event would also result in a larger s-value. However, this alternative explanation is unlikely. Using TpMglB-2WA as an example and assuming a monomer–dimer equilibrium for the protein when ligand is bound, the observed ΔsDSV and the concentrations used would indicate a KD of about 40 mM. However, if doubt exists, standard SV experiments with saturating concentrations of ligand and various concentrations of protein could be undertaken to corroborate the conformational-change hypothesis.
We have incorporated several aspects of our methodologies into a new freeware program called “DiSECT” (Difference Sedimentation to Elucidate Conformational Transitions). The program performs all of the calculations necessary to determine ΔsDSV. There are a number of built-in accessory functions also, including integration of the c(s) distribution from a SAM experiment, calculation of sav and σav, and determination of ΔsDSV expected from ligand-binding alone. It is critical to perform the latter calculation to correlate ΔsDSV with conformational changes as opposed to mere gains in mass from ligand binding. Functionality from HullRad (Fleming and Fleming 2018) is also included in DiSECT to enable the calculation of a predicted Δs from structural models of two conformational states. Any user may download the software from https://biophysics.swmed.edu/MBR/software.html.
Methods
Protein purification
Bovine serum albumen (BSA) was purchased from Sigma-Aldrich Corp. (St. Louis, MO; Cat#A7030). The powder was dissolved in phosphate-buffered saline buffer (PBS, 10 mM sodium phosphate, 1.76 mM potassium phosphate, 137 mM NaCl, 2.7 mM potassium chloride, pH 7.4) to a final concentration of 20 mg/mL, then filtered through a 0.22-μm centrifuge filter unit (Millipore). The solution was cooled to 4 °C and all subsequent purification steps occurred at this temperature. This solution was applied to a Superdex 200 16/60 size-exclusion column (GE Healthcare Bio-Sciences, Marlborough, MA) that had been equilibrated with PBS. Fractions deemed likely to contain the BSA monomer were pooled and concentrated to 8–10 mg/mL using a Millipore Ultra-4 centrifugation concentrator. Concentrations were determined spectrophotometrically using extinction coefficients calculated by the ProtParam utility of ExPASy (Gasteiger et al. 2005).
TpMglB-2WA was expressed and purified as described elsewhere (Brautigam et al. 2018). The composition of the protein’s storage buffer (Buffer B) was 10 mM sodium phosphate, pH 7.5, 100 mM NaCl.
Hydrodynamics calculations
HYDROPRO (de la Torre et al. 2000; Ortega et al. 2011) was used to calculate the hydrodynamic properties of the apo- and holo-TpMglB-2 models resulting from the crystallography (Brautigam et al. 2018). Masses of the polypeptides were calculated from the respective amino-acid compositions, as were the respective partial-specific volumes (Laue et al. 1992). The mass of d-glucose was included in the calculation for the holo-TpMglB-2 structure. Because there were slight differences in the termini of the proteins, the models were manually truncated such that the same number of amino-acid residues were present in both.
The same PDB files were used in the analysis by HullRad. We used a calculator embedded in DiSECT that used the HullRad code (Fleming and Fleming 2018) to calculate the hydrodynamic properties of the individual structures and report on their differences. Masses were automatically calculated form the contents of the PDB files.
Analytical ultracentrifugation
All AUC SV experiments were performed in an Optima XL-I analytical ultracentrifuge (Beckman-Coulter, Indianapolis, IN) at 20 °C using the Rayleigh interferometer only. Charcoal-filled Epon centerpieces were positioned between two sapphire windows in aluminum housings. In the SV method experiments, the reference sector was filled with Buffer B, and the sample sector was filled with 1 mg/mL TpMglB-2WA in Buffer B supplemented with either 1 mM d-ribose or 1 mM d-glucose. A total of six cells were prepared, three with d-ribose, and three with d-glucose. The assembled cells were then inserted into the holes of an An50-Ti rotor and incubated in the centrifuge under vacuum for ca. 2.5 h. Next, centrifugation was commenced at 50,000 rpm, with one scan collected every 1 min until no evidence of solute migration could be observed.
For DSV, we performed three simultaneous experiments: (1) a “SAM” experiment, in which Buffer B was introduced into the reference sector, and ~ 5 mg/mL protein was inserted into the sample sector; (2) a negative control, in which both sectors were filled with identically prepared solutions of protein with 1 mM ligand (d-glucose), and (3) the conformational change experiment, in which both sectors contained ca. 5 mg/mL protein, with the reference sector having 1 mM mock ligand (d-ribose) and the sample sector containing 1 mM d-glucose. In experiments (2) and (3), the sample sector was deliberately underfilled by ca. 6 μL, introducing a meniscus mismatch. This practice was suggested by Schachman and coworkers (Kirschner and Schachman 1971b) to yield more readily interpretable DSV patterns. The assembled cells were positioned in an An50-Ti rotor which was placed in the centrifuge and temperature-equilibrated under vacuum for at least 2.5 h. Centrifugation was then initiated at 50,000 rpm. One interferometric scan was collected every 1 or 0.5 min; a total of 999 scans was always collected.
The same procedures were used for the BSA-containing experiments, except as follows. The buffer used was PBS. Samples were supplemented to 4% (v/v) with either D2O (99.9%, Sigma Cat#151882) or H2O to mimic a conformational change in the protein.
Data analysis
All scans were time-stamp corrected (Zhao et al. 2013) using the software REDATE (https://biophysics.swmed.edu/MBR/software.html). The data were analyzed using SEDFIT (Schuck 2000), applying the following parameters during the c(s) analysis: Maximum Entropy regularization at a confidence level of 0.683, final s-resolution of 150, smin of 0.0 S, smax of 10.0 S, radially invariant and time-invariant noise calculation (Schuck and Demeler 1999). Integration (Schuck 1998) in SEDFIT was used to determine the best weighted-average s-value (sw,b) for each replicate. A multi-step method (Schuck 2016) to determine the error (σw,b) for each replicate was employed. First, F-statistics were used to determine the extreme values of the sample meniscus (m+ and m−) that were still compatible with the data (a confidence level of 68.3% was used). The distributions were integrated again at these extrema, and the resulting s-values were defined as sw,m+ and sw,m−. Then, with the menisci fixed at m+ and m−, two Monte Carlo (MC) simulations were carried out in SEDFIT to examine the error introduced by the integration and regularization. We found that the reported 68.3% confidence intervals from the MC simulations did not bracket the respective sw,m+ or sw,m−. However, we surmised that the simulations still captured the amount of error expected from integration and regularization, and thus we defined σw,m+ and σw,m− as one half of the reported interval from the Monte Carlo procedure. This resulted in four potential values to describe the confidence interval on sw,b: sw,m+ ± σw,m+ and sw,m− ± σw,m− We took the maximum and minimum (sw,max and sw,min, respectively) of these four to describe the error interval, and σw,b was then defined as the greater of sw,max − sw,b and sw,b − sw,min. Finally, the calculation of the weighted mean, sav, was accomplished via
and the overall σ for the respective sav, σav, was
where n is the number of replicates, which was always three in our studies.
To analyze the DSV experiments, the standard c(s) methodology was used to analyze the SAM experiment. The remaining analyses were accomplished in DiSECT. First, the experimental cell was identified by the user, and then the software loaded the last ten scans from that cell. It examined these and estimated the positions of the reference and sample menisci (mr and ms, respectively). At this point, the two menisci and the radial integration limits (rmin and rmax, i.e., the radii that all integrations must be within) were adjusted by the user if necessary. Next, the SAM-control c(s) distribution from SEDFIT was pasted into DiSECT’s integration tool, and the calculated values of s (sU) and the total fringe signal (ΔJU) were communicated by the tool to DiSECT. Finally, the method was initiated. All steps were automated from that point forward. The program calculated the time-invariant (TI) noise (Schuck and Demeler 1999) from the last ten scans and subtracted it from all subject scans. It located the identity of the scan in which the boundary should have roughly traversed half of the solution column by defining an integrated time (\(\int {\omega^{2} {\text{d}}t}\), which is recorded by the instrument in the header of the scan file) that met this criterion:
Having established which scan best meets this criterion, the algorithm moved backward through time, recording the scans less the TI noise and fitting a Gaussian curve to each. When the − 3σ (or − 4σ; this is user-adjustable) value of the curve was less than rmin, the search stopped and an analogous search in the forward direction in time was made. Thus, the algorithm automatically selected for the time range that would be included in the analysis. Next, the routine iterated through all included scans, establishing a separate baseline for each (this was necessary despite the subtraction of the TI noise). The y values of this baseline-corrected difference curve were termed ΔJ(r,rav)corr, with the mean radius of the DSV peak, rav, serving as a time variable. The first moment of the curve was found by multiplying ΔJ(r,rav)corr by r and performing a trapezoidal integration of the resulting curve; this was followed by normalization, yielding the ordinate (y) in the linear plot used to deduce Δs/\(\overline{s}\):
where rs, the solvent plateau, was the mean of the respective fitted Gaussian curve less 3 (or 4) times the respective sigma, and rp, the solution plateau, was the mean plus 3σ (or 4 σ), J0 was defined as ΔJU, and \(\overline{m}\) was the mean of the reference and sample menisci. The absolute value was taken so that a change in conformation would always result in a positive slope in the analyzed curve when mr < ms. For the abscissa, it was first necessary to calculate \(\overline{r}\):
The abscissa (x) was then calculated as
This version of the abscissa differs from that originally proposed by Kirschner and Schachman (Kirschner and Schachman 1971b). It is corrected for the fact that differences in the solute’s sedimentation velocity results in baseline offsets in the centrifugal portion of the DSV peak (Fig. 1) (Kirschner and Schachman 1971b; Skerrett 1975). A plot of y vs. x for all analyzed scans was fitted to a straight line, with the slope corresponding to Δs/\(\overline{s}\) and the y-intercept reporting on \(\left| {m_{{\text{s}}} - m_{{\text{r}}} } \right|\). Using the values obtained from the linear regression, a fit of baseline-subtracted interference data scans was initiated, with the starting value of Δs being Δslin:
Only the portions of the curves that were integrated in Eq. (4) were analyzed. To fit the difference curves directly, the concentration profiles for the reference (ΔJr(r,t,sr,D)) and sample sectors (ΔJs(r,t,ss,D)) were calculated separately, then values for the reference sector were subtracted from those for the sample sector, resulting in ΔΔJ(r,t,sr,ss,D). The equation used corresponded to the transport terms in the approximate analytic solution of the Lamm Equation formulated by Behlke and Ristau (2002), that is
where the symbol Φc denotes the complementary error function (sometimes called erfc) of the quantity enclosed in brackets, and using for convenience the dimensionless parameters:
and the auxiliary parameters
For the reference sector, \(s = s_{{\text{U}}}\) and \(m = m_{{\text{r}}}\); for the sample sector, \(s = s_{{\text{U}}} + \Delta s\) and \(m = m_{{\text{s}}}\). The diffusion coefficient (taken to be the same for both sectors) was a fitted parameter in this analysis; its initial value could be estimated from the molar mass of the solute using the Svedberg Equation, or it could simply be initialized at a realistic value (4–5 F in this paper), as we found that it had a robust radius of convergence. After ΔΔJ(r,t,sr,ss,D) was calculated, a straight line between its first and last values was subtracted from it to mimic the baseline subtraction that was performed for the raw data. It was this function that was fitted to the baseline-subtracted interferometric data using the minimization algorithm of Levenberg and Marquardt (1963) and the Nelder–Mead simplex algorithm (Nelder and Mead 1964). The above operations were typically performed on 80–200 scans in just a few seconds using the program.
Data simulations
The simulated DSV data were generated using a combination of SEDFIT and a custom-written Python script. First, two standard SV data sets (one each for the reference and sample sectors) were simulated using typical parameters in SEDFIT; random Gaussian noise was added to each data set at a level of 0.005 fringes. Care was taken to build in a meniscus offset and to ensure that both data sets all had perfectly paired timestamps in their respective headers. The data sets were written to separate file folders. Then, the script was employed to pairwise subtract the reference sector data from the sample-sector data, and the results were written as new files into a new file folder. These data could be read by DiSECT and subjected to the algorithms described above.
Availability of data and material
Available upon request.
References
Bai X, Fernandez IS, Mcmullan G, Scheres SHW (2013) Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. Elife 2:e00461
Behlke J, Ristau O (2002) A new approximate whole boundary solution of the Lamm differential equation for the analysis of sedimentation velocity experiments. Biophys Chem 95:59–68
Bennett WS, Steitz TA (1978) Glucose-induced conformational change in yeast hexokinase. Proc Natl Acad Sci USA 75:4848–4852
Borges JC, Pereira JH, Vasconcelos IB et al (2006) Phosphate closes the solution structure of the 5-enolpyruvylshikimate-3-phosphate synthase (EPSPS) from Mycobacterium tuberculosis. Arch Biochem Biophys 452:156–164
Borrok MJ, Kiessling LL, Forest KT (2007) Conformational changes of glucose/galactose-binding protein illuminated by open, unliganded, and ultra-high-resolution ligand-bound structures. Protein Sci 16:1032–1041
Borrok MJ, Zhu Y, Forest KT, Kiessling LL (2009) Structure-based design of a periplasmic binding protein antagonist that prevents domain closure. ACS Chem Biol 4:447–456
Brautigam CA, Deka RK, Liu WZ, Norgard MV (2018) Crystal structures of MglB-2 (TP0684), a topologically variant d-glucose-binding protein from Treponema pallidum, reveal a ligand-induced conformational change. Protein Sci 27:880–885
Brautigam CA, Deka RK, Liu WZ, Norgard MV (2016) The Tp0684 (MglB-2) lipoprotein of Treponema pallidum: a glucose-binding protein with divergent topology. PLoS ONE 11:e0161022
Brown PH, Balbo A, Schuck P (2007) Using prior knowledge in the determination of macromolecular size-distributions by analytical ultracentrifugation. Biomacromol 8:2011–2024
Brown PH, Schuck P (2008) A new adaptive grid-size algorithm for the simulation of sedimentation velocity profiles in analytical ultracentrifugation. Comput Phys Commun 178:105–120
Catterall WA, Wisedchaisri G, Zheng N (2017) The chemical basis for electrical signaling. Nat Chem Biol 13:455–463
Davidson AL, Maloney PC (2007) ABC transporters: how small machines do a big job. Trends Microbiol 15:448–455
de la Torre JG, Huertas ML, Carrasco B (2000) Calculation of hydrodynamic properties of globular proteins from their atomic-level structures. Biophys J 78:719–730
Desai A, Krynitsky J, Pohida TJ et al (2016) 3-D printing for analytical ultracentrifugation. PLoS ONE 11:e0155201
Deupi X, Standfuss J (2011) Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr Opin Struct Biol 21:541–551
Dwyer MA, Hellinga HW (2004) Periplasmic binding proteins: a versatile superfamily for protein engineering. Curr Opin Struct Biol 14:495–504
Errington N, Rowe AJ (2003) Probing conformation and conformational change in proteins is optimally undertaken in relative mode. Eur Biophys J 32:511–517. https://doi.org/10.1007/s00249-003-0315-x
Fawaz MV, Topper ME, Firestine SM (2011) The ATP-grasp enzymes. Bioorg Chem 39:185–191
Felder CB, Graul RC, Lee AY et al (1999) The venus flytrap of periplasmic binding proteins: an ancient protein module present in multiple drug receptors. AAPS J 1:7–26
Fleming PJ, Fleming KG (2018) HullRad: Fast calculations of folded and disordered protein and nucleic acid hydrodynamic properties. Biophys J 114:856–869
Fresquet M, Jowitt TA, Ylöstalo J et al (2007) Structural and functional characterization of recombinant matrilin-3 A-domain and implications for human genetic bone diseases. J Biol Chem 282:34634–34643
Gasteiger E, Hoogland C, Gattiker A et al (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The proteomics protocols handbook. Humana Press, Totowa, pp 571–607
Gerhart JC, Schachman HK (1968) Allosteric interactions in aspartate transcarbamylase. II. Evidence for different conformational states of the protein in the presence and absence of specific ligands. Biochemistry 7:538–552
Gutmann T, Kim KH, Grzybek M et al (2018) Visualization of ligand-induced transmembrane signaling in the full-length human insulin receptor. J Cell Biol 217:1643–1649
Harris SJ, Winzor DJ (1988) Thermodynamic nonideality as a probe of allosteric mechanisms: preexistence of the isomerization equilibrium for rabbit muscle pyruvate kinase. Arch Biochem Biophys 265:458–465
Jacobsen MP, Winzor DJ (1997) Studies of ligand-mediated conformational changes in enzymes by difference sedimentation velocity in the Optima XL-A ultracentrifuge. Prog Colloid Polym Sci 107:82–87
Jeschke G (2012) DEER distance measurements on proteins. Annu Rev Phys Chem 63:419–446
Kamata K, Mitsuya M, Nishimura T et al (2004) Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure 12:429–438
Khan FI, Lan D, Durrani R et al (2017) The lid domain in lipases: structural and functional determinant of enzymatic properties. Front Bioeng Biotechnol 5:1–13
Kirschner MW, Schachman HK (1971a) Conformational changes in proteins as measured by difference seimentation studies. II. Effect of stereospecific ligands on the catalytic subunit of aspartate transcarbamylase. Biochemistry 10:1919–1926
Kirschner MW, Schachman HK (1971b) Conformational changes in proteins as measured by difference sedimenation studies. I. A technique for measuring small changes in sedimenation coefficient. Biochemistry 10:1900–1919
Kornblatt JA, Schuck P (2005) Influence of temperature on the conformation of canine plasminogen: an analytical ultracentrifugation and dynamic light scattering study. Biochemistry 44:13122–13131. https://doi.org/10.1021/bi050895y
Krishnan N, Koveal D, Miller DH et al (2014) Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat Chem Biol 10:558–566
Länge K, Rapp BE, Rapp M (2008) Surface acoustic wave biosensors: a review. Anal Bioanal Chem 392:1509–1519
Laue TM, Shah BD, Ridgeway RM, Pelletier SL (1992) Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC (eds) Analytical ultracentrifugation in biochemistry and polymer science. The Royal Society of Chemistry, Cambridge, pp 90–125
Li X, Mooney P, Zheng S et al (2013) Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods 10:584–590. https://doi.org/10.1038/nmeth.2472
Mao B, Pear MR, McCammon JA, Quiocho FA (1982) Hinge-bending in l-arabinose- binding protein: the “Venus-flytrap” model. J Biol Chem 257:1131–1133
Marquardt DW (1963) An algorithm for the least-squares estimation of nonlinear parameters. J Soc Ind Appl Math 11:431–441
Moree B, Connell K, Mortensen RB et al (2015) Protein conformational changes are detected and resolved site specifically by second-harmonic generation. Biophys J 109:806–815
Nelder J, Mead R (1964) A simplex method for function minimization. Comput J 7:308–313
Newcomer ME, Lewis BA, Quiocho FA (1981) The radius of gyration of l-arabinose-binding protein decreases upon binding of ligand. J Biol Chem 256:13218–13222
Oberfelder RW, Consler TG, Lee JC (1985) Measurement of changes of hydrodynamic properties by sedimentation. Methods Enzymol 117:27–40
Oberfelder RW, Lee LL-Y, Lee JC (1984) Thermodynamic linkages in rabbit pyruvate kinase: kinetic, equilibrium, and structural studies. Biochemistry 23:3813–3821
Ortega A, Amorós D, García De La Torre J (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys J 101:892–898
Persons JD, Khan SN, Ishima R (2018) An NMR strategy to detect conformational differences in a protein complexed with highly analogous inhibitors in solution. Methods 148:9–18
Philo JS (1997) An improved function for fitting sedimentation velocity data for low- molecular-weight solutes. Biophys J 72:435–444
Rees AW, DeBuysere MS, Lewis EA (1977) Difference sedimentation velocity adapted to low molecular weight proteins. Arch Biochem Biophys 182:478–487
Richards EG, Schachman HK (1959) Ultracentrifuge studies with Rayleigh interference optics. I. General applications. J Phys Chem 63:1578–1591
Schuck P (2000) Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J 78:1606–1619
Schuck P (2016) Sedimentation velocity analytical ultracentrifugation: discrete species and size-distributions of macromolecules and particles. CRC Press, Boca Raton
Schuck P (1998) Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys J 75:1503–1512
Schuck P, Demeler B (1999) Direct sedimentation analysis of interference optical data in analytical ultracentrifugation. Biophys J 76:2288–2296
Shang G, Zhang C, Chen ZJ et al (2019) Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature 567:389–393
Singh D, Tamao Y, Blakley RL (1977) Regulation and cooperativity: the case of ribonucleotide reductase of Lactobacillus leischmanii. Adv Enzyme Regul 15:81–99
Skerrett RJ (1975) The theory of the difference sedimentation method. Anal Biochem 66:1–11
To SC, Brautigam CA, Chaturvedi SK et al (2019) Enhanced sample handling for analytical ultracentrifugation with 3D-printed centerpieces. Anal Chem 91:5866–5873
Uchikawa E, Choi E, Shang G et al (2019) Activation mechanism of the insulin receptor revealed by cryo-EM structure of the fully liganded receptor–ligand complex. Elife 8:e48630
Yikilmaz E, Rouault TA, Schuck P (2005) Self-association and ligand induced conformational changes of iron regulatory proteins 1 and 2. Biochemistry 44:8470–8478
Zhao H, Ghirlando R, Piszczek G et al (2013) Recorded scan times can limit the accuracy of sedimentation coefficients in analytical ultracentrifugation. Anal Biochem 437:104–108
Acknowledgements
The authors wish to thank Dr. Peter Schuck for helpful discussions and the use of one of the analytical ultracentrifuges in his lab to collect some of the data presented herein. Parts of this research were supported by a Grant (AI056305) to M.V.N. from the National Institutes of Health.
Funding
NIH Grant No. AI056305 to M.V.N.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Code availability
Compiled software freely available. Custom Python script available upon request.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Special Issue: Analytical Ultracentrifugation 2019.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Brautigam, C.A., Tso, SC., Deka, R.K. et al. Using modern approaches to sedimentation velocity to detect conformational changes in proteins. Eur Biophys J 49, 729–743 (2020). https://doi.org/10.1007/s00249-020-01453-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-020-01453-w