
Abstract
The membrane-association properties of the amino-terminal domain of huntingtin are accompanied by subcellular redistribution of the protein in cellular compartments. In this study we used tryptophan substitution of amino-acid residues at different positions of the huntingtin 1–17 domain (Htt17) to precisely determine, for the first time, the depth of penetration of the peptides within the lipid bilayer. Initially, secondary structure preferences and membrane association properties were quantitatively determined for several membrane lipid compositions; they were found to be closely related to those of the natural peptide, indicating that changes in the sequence had little effect on these characteristics of the domain. The tryptophan-substituted peptides became inserted into the membranes’ interfacial region, with average tryptophan positions between 7.5 and 11 Å from the bilayer center, in agreement with in-plane orientation of the peptide. Participation of the very-amino terminus of the peptide in the membrane-association process was demonstrated. The results not only revealed the occurrence of association intermediates when the huntingtin 1–17 anchoring sequence became inserted into the membrane but also suggest the formation of aggregates and/or oligomers during membrane association. When inserted, the F11W site was of crucial importance in lipid anchoring and stabilization of the whole peptide, whereas the terminal residues are located close to the membrane surface. The carboxy-terminal tryptophan (F17W), which also constitutes the site of the polyglutamine extension in the natural domain, was found closest to the aqueous environment, accompanied with the highest aqueous quenching constants. These results were used to propose a refined model of lipid interactions of the huntingtin 1–17 domain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Huntington’s disease is a neurodegenerative disorder involving coordinative malfunction and cognitive restriction. The codon expansion of more than 37–40 CAG repeats of the mutated, causative huntingtin gene leads to an enlarged polyglutamine (polyQ) tract of the corresponding protein product which interferes with the native functions of this protein. The variety of functions of huntingtin protein in healthy cells includes calcium homeostasis, axonal transport, and subcellular localization (Rockabrand et al. 2007; Orr et al. 2008; Kegel et al. 2005). On polyQ expansion, such as occurs in the disease, aggregation and fibril formation lead to induction of the death of neuronal cells, in particular, finally resulting in loss of cellular mass in the striatum of the central nervous system.
The amino terminus of huntingtin encompasses 17 amino-acid residues (Htt17) directly adjoined to the polyQ tract. This small domain is of crucial importance in modulation of the fibrillation process of polyQ proteins. Htt17 undergoes conformational transitions from a random coil in aqueous environments to high α-helix content on polar interaction with polyQ peptides, strongly enhancing polyQ oligomerization (Tam et al. 2006). The amphipathic character of this α-helix induces accumulation of the Htt17 domain by nonpolar clustering, which brings together several polyQ tracts, which are believed to initiate oligomerization and fibrillization (Thakur et al. 2009; Tam et al. 2009).
Huntingtin has been described as being located in the nucleus, ER, Golgi apparatus, and mitochondria (Rockabrand et al. 2007; Atwal et al. 2007). Atwal et al. (2007) were the first to show the lipid-binding properties of Htt17, and their direct involvement with vesicle association and the cross-talk between nucleus and ER. Mutation of the Htt17 domain (M8P) led to accumulation inside the nucleus accompanied by cellular toxicity. Further biophysical analysis of the lipid-binding properties of isolated Htt17 peptides revealed lipid-binding specificity, with apparent dissociation constants in the range of those of antimicrobial peptides, and conformational transitions from random coil to α-helical secondary structures (Michalek et al. 2013a). Solid-state NMR measurements revealed that when interacting with supported phospholipid bilayers the α-helical Htt17 peptide became aligned approximately parallel to the plane of the lipid bilayer (Michalek et al. 2013a). The structure of Htt17 in DPC micelles and its approximate location along the micellar interface was determined by solution NMR (pdb: 2LD2). In combination with orientational restraints obtained from solid-state NMR measurements on oriented bilayer samples the structure was refined and the detailed tilt and pitch angles of the Htt17 helix were determined (Michalek et al. 2013a). The amino-terminal domain is oriented at an angle of approximately 13° to the membrane surface, which ensures that the polar face of the amphipathic helix is directed toward the hydrophilic environment. This topology of Htt17 favors its interactions with polyQ tracts, which correlates with its known function in modulating the oligomerization of polyQ proteins (Tam et al. 2006, 2009; Thakur et al. 2009).
Previous studies have revealed the lipid-binding properties of amino-acid residues 6–17 of Htt17. This prompted us to analyze the lipid-binding capacity of other parts of the primary structure by tryptophan emission fluorescence spectroscopy using tryptophan-substituted Htt17 derivatives. Because previous mutational analysis of the Htt17 peptide (M8P) affected its lipid-binding properties (Atwal et al. 2007), positions of the peptide were modified which did not alter either its polar character or the hydrophobicity of the amino-acid residues. As a result, participation of the very first amino-terminal amino-acid residues in lipid interactions was revealed. The different lipid-binding properties and penetration depths of the Htt-W derivatives enabled us to propose a refined model of the membrane-anchoring interactions of huntingtin by the Htt17 domain.
Materials and methods
Materials
All lipids were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Oligonucleotides were synthesized by MWG (Ebersberg, Germany).
Construction of expression vectors
The codon-optimized gene sequence (Geneart, Regensburg, Germany) of Htt17 was cloned into the pTIPX-4 expression vector containing a kanamycin resistance gene by use of BamHI and SacI restriction cleavage sites. For construction of tryptophan mutants of Htt17, site-directed mutagenesis PCR reactions were performed by use of the oligonucleotides: M1W forward, GAATTGGATCCGTGGGCGACCCTGGAAAAACTG; reverse, GGCCGAGCTCTTATTAGAAGCTTTTCAGGCTTCGAA; F11W forward, GAATTGGATCCGATGGCGACCCTGGAAAAACTG; reverse, GGCCGAGCT CTTATTAGAAGCTTTTCAGGCTTTCCCACGCTTTCATCAG; F17W forward, GAATTGGATCCGATGGCGACCCTGGAAAAACTG; reverse, GGCCGAGCTCTTATTACCAGCTTTTCAGGCTTTCGAA. For all plasmids, correct insertion was verified by sequencing (MWG). Because of the cloning sites used for expressing the peptides in the pTIPX4 vector, all peptides contained an additional proline at the amino-terminus.
Recombinant expression and purification of Htt17 tryptophan mutants
The recombinant expression of Htt17 peptides was performed after transforming the pTIPX4-Htt17 plasmid into E. coli BL21 DE3 (Invitrogen, Cergy Pontoise, France). Bacterial cultures were grown in 1 L LB medium containing 50 µg/mL kanamycin at 37 °C and 200 rpm until the optical density at 600 nm (OD600) was 0.8. Over expression was then induced by addition of 1 mM IPTG and additional growth for 3 h.
The TAF12-Htt17 fusion protein was purified by centrifugation of the bacterial suspension at 3000g for 30 min at 4 °C, followed by resuspension in 50 mM Tris–HCl, 100 mM NaCl, 5 mM EDTA, 0.5 % (v/v) Triton X-100, pH 8.0. The suspension was sonicated for 5 min at 25 % power on a Bandelin Sonoplus HD 200 and subsequently centrifuged at 10,000g for 30 min at 4 °C. The insoluble and soluble parts were investigated by SDS-PAGE analysis. All fusion proteins were found in the insoluble part, as a result of aggregation of inclusion bodies during expression. Inclusion bodies were resolved in 75 % (v/v) formic acid and incubated for 24 h at 50 °C followed by evaporation of the acid under vacuum. Samples were resuspended in distilled, sterilized water and loaded on to a Prontosil C4-HPLC column (Bischoff, Leonberg, Germany). RP-HPLC with an acetonitrile–water gradient was used for purification; Htt17 derivatives eluted at ~31 % (v/v) MeCN–water containing 0.1 % TFA. All peptides were exposed to HFIP–TFA for disaggregation, as described elsewhere (Chen and Wetzel 2001), and TFA counter ions were exchanged twice in 5 % (v/v) acidic acid–water before functional assays. The exact theoretical masses of purified peptides (>90 %) were verified by MALDI-TOF-mass spectrometry with M1W: 2,126.2 (±1) m/z; F11W: 2,109.8 (±1) m/z; F17W: 2,109.8 (±1) m/z.
Size-exclusion chromatography
Molecular size-exclusion chromatography was performed on a Biologic Duoflow FPLC system (Biorad, CA, USA) equipped with a HiPrep 16/60 Sephacryl S-100 high-resolution column (GE Healthcare Bio-Sciences, Uppsala, Sweden) to analyze the oligomeric states of the samples. Peptides were separated in 20 mM Tris–HCl, 75 mM NaCl pH 7.3 at a flow of 1 mL/min at 4 °C; the detection wavelength was 214 nm. For calibration, ribonuclease A (13,700 Da), carbonic anhydrase (29,000 Da), and bovine serum albumin (66,000 Da) were used to generate the standard slope by plotting K av against log MW, as described in Gel Filtration: Principles and Methods (http://www.gelifesciences.com/protein-purification).
Vesicle preparation
For vesicle preparation, lipid mixtures comprising POPC–POPS (75:25 % mol/mol), POPE–POPG (75:25 % mol/mol), POPC (100 %), and POPC–POPS–cholesterol (45:15:40 % mol/mol) were dissolved in chloroform and subsequently evaporated under a nitrogen stream to obtain a thin lipid film. Residual solvent was removed by lyophilization, followed by rehydration for 1.5 h by continuous vortex mixing in 20 mM Tris–HCl, 75 mM NaCl pH 7.3. The suspension was subjected to three cycles of freezing in liquid nitrogen and thawing in a water bath at 37 °C. Finally, small and large unilamellar vesicles (SUV, LUV) were produced by extrusion 21 times through 50 or 100 nm polycarbonate membranes (Avestin, Mannheim, Germany), respectively.
Circular dichroism spectroscopy
Circular dichroism spectroscopy was performed in 50 mM sodium phosphate buffer, 75 mM sodium chloride, pH 7.3, on a Jasco (Tokyo, Japan) J-810 CD spectrometer with a bandwidth of 1 nm and a cuvette of 1 mm path length. The peptide concentrations were 36 µM and POPC–POPS 75:25 (mol/mol) SUVs were added to a P/L ratio of 1:30 (mol/mol). The dilution factor was recalculated after each experiment. Four scans with a scan speed of 100 nm/min were averaged and the CD signal of the buffer was subtracted subsequently. Analysis of spectra was performed using Origin 6.0 and helical contents were calculated by use of the CDSSTR algorithm (Sreerama and Woody 2000).
Tryptophan-emission fluorescence
Tryptophan-emission fluorescence measurements were performed on a Fluorolog fluorescence spectrometer (Horiba Jobin–Yvon, NJ, USA) with an excitation wavelength of 290 nm at 20 °C, when not indicated otherwise. LUV lipid mixtures were added from stock solutions of 5 mg/mL to solutions of Htt17-M1W, Htt17-F11W, or Htt17-F17W (32 µM final concentration) in 20 mM Tris–HCl, 75 mM NaCl, pH 7.3, with continuous stirring, in quartz cuvettes of 1 cm path length (Hellma Analytics, Müllheim, Germany). Emission and excitation band passes were set to 4 nm and emission spectra were recorded, after short incubation, from 300 to 450 nm, averaging 3 scans. Spectra were corrected for light scattering, baseline, and dilution after adding lipid vesicles for each titration step. Blue shifts were calculated from the emission maxima of peptide and peptide–lipid mixtures. The standard deviation of the blue shift was 1 nm.
The binding of Htt17-W derivatives was described by use of the equations below, where Θ is the fraction of bound peptide, derived from previous observations (Dubreil et al. 1997).
The apparent dissociation constant (K d) was determined by plotting the measured wavelength (λ) for each titration step of added lipid concentration (L), with n being the Hill coefficient and [λmax] or [λmin] being the maximum wavelength shift with and the minimum without lipids, respectively. The data for tryptophan emission were fit sigmoidally by use of Origin 6.0.
Calculation of differential binding isotherms
The different dissociation constants obtained for M1W, F11W, and F17W suggest that the peptide associates with the membrane in succinct steps. Therefore, populations of intermediate binding states were calculated from normalized binding isotherms of individual tryptophan replacements (M1W, F11W, and F17W). By taking differences of the binding isotherms two intermediates were considered. The fraction of a first intermediate was obtained by considering the differences between the association of M1W and F17W: \(f_{\text{int}}^{1} = f^{M1W} - f^{F17W}\). Furthermore \(f_{\text{int}}^{2} = f^{F17W} - f^{F11W}\) is the fraction of peptide with F17W but not F11W bound and/or inserted. Thus, for \(K_{d}^{F11W} > K_{d}^{f17W} > K_{d}^{M1W}\) \(f_{\text{int}}^{1}\) is a state where only residue 1 is exposed to a hydrophobic environment whereas \(f_{\text{int}}^{2}\) is a state where residues 1 and 17 are exposed to a hydrophobic environment.
Acrylamide quenching
Quenching of tryptophan emission by water-soluble acrylamide was detected at an excitation wavelength of 290 nm, with the same settings as described above. Acrylamide stock solution (5.6 M) was added stepwise to 32 µM peptide solutions in the absence and presence of 1.8 mM LUV lipid mixtures. Spectra were corrected for dilution and for baselines obtained with no peptides present. Integrals of the emission spectra were analyzed by use of the Stern–Vollmer equation (Chang and Ludescher 1994):
where F 0 and F were, respectively, the fluorescence intensities in the absence and in the presence of quencher at a concentration [Q], and V is a static quencher constant. The Stern–Vollmer quenching constant K sv facilitates comparison of the quencher’s accessibility to the tryptophan residues in aqueous solution or membrane environment.
Red-edge excitation shift (REES)
Tryptophan residues in aqueous and lipid environments were analyzed by red-edge excitation shift with stepwise variation of the excitation wavelength from 280 to 315 nm for peptide samples (Htt17-M1W, Htt17-F11W, and Htt17-F17W) at a concentration of 32 µM in the absence and presence of 1.8 mM LUV lipid mixtures at 20 °C, with the same settings as described above. The intensities of spectra were corrected for baseline (solution without peptide) and the emission maxima were plotted against excitation wavelength. A red shift of the emission maxima as a result of the shift of the excitation wavelength is caused by slow solvent relaxation around the excited state of tryptophan (Demchenko and Ladokhin 1988; Ghosh et al. 1997).
Collisional quenching by dibrominated phosphatidylcholine
Phosphatidylcholine brominated at defined positions of the acyl chain has been shown to cause only minor perturbations of the membrane and was therefore used to analyze the depth of tryptophan insertion into the lipid bilayer (Bolen and Holloway 1990). LUVs containing POPC–POPS (75:25 mol/mol) plus 30 % diBr-POPC (0.9 mM final concentration) were added to 16 µM peptide solutions in 20 mM Tris–HCl, 75 mM NaCl, pH 7.3, and incubated for 2 h at 20 °C. Emission spectra from 300 to 400 nm were recorded and averaged from 3 scans. Spectra were corrected for the baseline (solution in the absence of peptide) and the integrals of the intensity spectra were taken to analyze quenching effects. The depth of tryptophan insertion was calculated by fitting the data by use of the distribution analysis method (Ladokhin et al. 1993):
where F 0 and F h represent the intensities in the absence and presence of quencher (h), and c(h) is the concentration of the quencher (which is equal for all the brominated lipids used). S is the area under the curve (effectiveness of quenching), σ is the dispersion of the depth in the bilayer, h m is the distance of tryptophan from the center of the bilayer, and h is the distance of the bromine from the center of the bilayer (10.8, 8.3, and 6.3 Å for dibromo-(6,7)-POPC, (9,10)-POPC, and (11,12)-POPC, respectively) (McIntosh and Holloway 1987).
Results
The theoretically calculated mean hydrophobicity of the chemically synthesized peptide Htt17-syn (MATLEKLMKAFESLKSF) is −0.08, which is similar to those of the recombinantly expressed tryptophan-substituted Htt17 derivatives Htt17-M1W (M1W: PWATLEKLMKAFESLKSF), Htt17-F11W (F11W: PMATLEKLMKAWESLKSF), and Htt17-F17W (F17W: PMATLEKLMKAFESLKSW), for which values are −0.07, −0.09, and −0.09, respectively (Eisenberg et al. 1984). To simplify comparison with the native sequences the numbering was maintained which brings the proline to position 0. In previous structural and functional studies the native and the P-Htt17 peptides had very similar lipid-binding properties (Michalek et al. 2013a, b), a finding which we confirmed by comparing the membrane association of a synthetic peptide Htt17-M1W in the presence or absence of the additional proline residue (not shown). The helical wheel projection for the α-helix of residues 6–17 shows the relative positions of tryptophan substitutions 11 and 17 (Fig. 1a).

Helical wheel projection and molecular characterization of the Htt17 peptides. a Htt17 adopts α-helical contents from residues 6–17 by interaction with lipid membranes (pdb: 2LD2). Arrows denote tryptophan substitutions in the α-helix for the derivatives used in this study. b The three tryptophan-substituted peptides, M1W (open diamond), F11W (open triangle), and F17W (open inverted triangle) were subjected to molecular size-exclusion chromatography at 4 °C after disaggregation. The K av values of the elution times were plotted against log MW to calculate the hydrodynamic molecular weights compared with the chemically synthesized Htt17 peptide (Htt17-syn, open circle). The extrapolated linear calibration slope is shown as a dashed line derived from the K av values of the calibration substances (filled squares). c CD spectra of 36 µM M1W, F11W, and F17W peptides, and natural Htt17, in buffer and in the presence of 1 mM POPC–POPS 75:25 mol/mol SUVs at 20 °C
Characterization of recombinantly expressed tryptophan-substituted Htt17 peptides
Purified recombinantly expressed Htt17-W-derivatives, M1W, F11W, and F17W, were subjected to size-exclusion chromatography to investigate the peptides’ oligomeric states in aqueous solution (Fig. 1b). F11W and F17W had approximately identical retention times compared with the synthesized Htt17 peptide (Htt17-syn) indicating a monomeric state in aqueous environments, in agreement with previous observations (Thakur et al. 2009). Although M1W had a longer retention time, still in good agreement with the monomeric state, interaction with the matrix of the size-exclusion column and minimized molecular volumes of the random coil overall structure reasonably explain the elution profile of this peptide.
Furthermore, the overall secondary structures of the Htt17-W-derivatives were inspected by CD spectroscopy and compared with the natural peptide (Fig. 1c). The CD spectra of the natural Htt17 sequence, M1W, F11W, and F17W differed only slightly, and all had predominantly random coil secondary structure in aqueous solution (Atwal et al. 2007; Michalek et al. 2013a). On addition of 1 mM POPC–POPS 75:25 (mol/mol) SUVs M1W, F11W, and F17W underwent a transition to α-helical secondary structures with contents of 67, 64, and 60 %, respectively. These values are in good agreement with previously reported values for chemically synthesized Htt17 in the presence of lipid vesicles (Michalek et al. 2013a).
Intrinsic tryptophan fluorescence emission measurements
To analyze the interactions of Htt17-W-derivatives with lipid vesicles of different composition, the shifts and intensities of intrinsic tryptophan emission fluorescence were measured. The first purpose of these experiments was to show that neither tryptophan substitution nor N-terminal proline addition substantially modified membrane association of the Htt17 domain. Second, fluorescence spectroscopic analysis revealed additional details of the membrane-association process that could not be observed by CD spectroscopy (Michalek et al. 2013a). Because it has previously been described for many lipid-interacting proteins and peptides, the shift to smaller wavelength (blue shift) and increased intensities of the tryptophan fluorescence emission originates from the change of polarity of the indole rings’ microenvironment caused by transfer of the tryptophan side chain from aqueous solution to the lipid bilayer.
M1W, F11W, and F17W were successively incubated with increasing concentrations of LUVs composed of POPC–POPS 75:25 (mol/mol), POPC–POPS–cholesterol 45:15:25 (mol/mol/mol), POPE–POPG 75:25 mol/mol, or pure POPC, and the emission fluorescence maximum or intensity increase at each step were plotted against lipid concentration (Fig. 2).

Intrinsic tryptophan fluorescence emission spectra of Htt17-W derivatives in the presence of increasing concentrations of different lipid LUVs. The shifts in maximum wavelengths (a–c) and the increases in the maximum intensities at 348, 340, and 355 nm (d–f) for M1W (a, d), F11W (b, e), and F17W (c, f) in buffer and with stepwise increases in the concentrations of LUVs composed of POPC–POPS 75:25 mol/mol (filled circles), POPC–POPS–cholesterol 45:15:25 mol/mol/mol (open squares), POPE–POPG 75:25 mol/mol (open triangles) and pure POPC (filled inverted triangles) were measured at 20 °C. The corresponding curve fitting was performed as described in the “Materials and methods” section
The maxima of the fluorescence emission in buffer were at wavelengths (λmax) 359, 359, and 365 nm for M1W, F11W, and F17W (Fig. 2a–c), respectively, reflecting exposure of the tryptophan residues to the aqueous environments (Pan et al. 2006). In the presence of 1.8 mM POPC–POPS, resembling model membranes of vesicles from the cytoplasm, the highest blue shifts and intensity increases (Fig. 2d–f) were observed when following λmax for M1W, F11W, and F17W at 348, 337, and 355 nm, respectively (Table 1). Incorporation of 40 mol % cholesterol into POPC–POPS vesicles, mimicking the lipid composition of the outer cytoplasmic membrane, affected the binding concomitant with a slower decrease in λmax on lipid titration. However, the fluorescence intensity of the peptides in the presence of cholesterol-containing vesicles increased only slightly for M1W and F11W and even decreased for F17W. In general, the intensity changes were indicative of multi-phase behavior in which, during the titration with lipids, an initial increase in intensity is followed by reduction of fluorescence at lipid concentrations >1 mM (Fig. 2d–f). This observation was suggestive of rearrangements in the conformation and/or oligomeric structure of the peptides leading to self-quenching of closely positioned tryptophan residues (cf. also below). Furthermore, residual scattering artifacts, that are not completely accounted for by the correction, could contribute to the decrease in fluorescence in the presence of high vesicle concentrations.
Addition of POPE–POPG, a model system for mitochondrial membranes, or zwitterionic POPC vesicles had, in general, a less pronounced effect on λmax or the fluorescence intensities. Titration of POPC vesicles resulted in a less complete binding at 1.8 mM lipid, in particular for F11W and F17W (Fig. 2b, c); a comparatively large fluorescence intensity increase was, nevertheless, observed for F17W (Fig. 2f). Indeed, visual inspection of Fig. 2 indicates that in several of the titration experiments even at the highest lipid concentrations only a fraction of the peptides is membrane-associated. However, it seems from visual extrapolation that for all membranes investigated the λmax of bilayer-associated peptides approaches values in the range of those obtained for POPC–POPS membranes; this value was therefore used for calculation of dissociation constants (Fig. 2). These results suggest a major electrostatic contribution to peptide–lipid interactions in which F11W inserts deeply into the hydrophobic environment. In contrast the M1W and F17W remained more exposed to the solvent.
For determination of apparent dissociation constants for the peptide–lipid interactions, λmax was plotted against the lipid concentration and analyzed quantitatively. The non-ideal behavior in the association was taken into account by introducing Hill coefficients into the fitting procedure (Table 1). Whereas the Hill coefficient is often believed to represent cooperative binding, it was originally introduced as an empirical approach to fit more complex binding isotherms without an underlying molecular model (Hill 1910). Here the lipid was titrated into a peptide solution, and the coefficient enables correction for a variety of effects that make the binding curve appear sigmoidal (Hill 1910; Mosior and McLaughlin 1991; Rhoades et al. 2006). Among possible reasons, the initial steps during the titration experiment are characterized by high peptide-to-lipid ratios and the membrane may be disrupted. Furthermore, excess charges at the lipid membrane, as a result of the presence of either acidic phospholipids or cationic peptides, cause accumulation or depletion of peptide in the vicinity of the membrane surface (Mosior and McLaughlin 1992; Wieprecht et al. 2000; Bechinger et al. 2004). The local peptide concentration does not, therefore, correspond to that of the bulk solution and, in addition, this redistribution changes during the titration process. Whereas this latter effect is best treated by Gouy–Chapman theory (Macdonald and Seelig 1988) this can be ambiguous when multiple non-idealities overlap or the experiments have not been optimized for this purpose.
For all three Htt-17 tryptophan derivatives the binding affinities toward zwitterionic POPC vesicles were lowest, with K d values of 132, 548, and 1,058 µM for the M1W, F11W, and F17W mutants, respectively (Table 1). Conversely, incorporation of POPS into mixed vesicles caused higher affinity with apparent dissociation constants of 21, 128, and 86 µM. Membrane-association decreased by a factor of approximately two to four on incorporation of 40 % cholesterol into the lipid mixtures. Pronounced differences in the binding affinities were observed in presence of POPE–POPG vesicles. Compared with POPC–POPS vesicles, reduced affinity was observed for M1W and F17W whereas the binding of F11W was similar, with a K d value of 135 µM (Table 1). These values and tendencies were in good agreement with the membrane dissociation constants that had been obtained by following the changes in peptide secondary structure on titration of POPC–POPS 75:25 (42 µM) or POPC SUVs (1,250 µM) using CD spectroscopy (Michalek et al. 2013a). The quantitative agreement is even better when it is taken into consideration that these previous studies were analyzed assuming a Hill coefficient of unity (Atwal et al. 2007), which for the data shown in Fig. 2 results in dissociation constants that are increased up to fourfold compared with Table 1 (not shown). From the apparent dissociation constants it becomes obvious that for many of the titration experiments shown in Fig. 2 significant amounts of peptide remain in aqueous solution even in the presence of 1.8 mM lipid vesicles.
The different dissociation constants obtained for M1W, F11W, and F17W can be explained on the basis of intermediates formed during binding (Fig. 5a–d). A first intermediate can be defined by the different binding between M1W and F17W. Whereas for POPC–POPS and POPE–POPG vesicles the presence of an intermediate state is clearly visible (open spheres) those intermediates are not as obvious in the presence of cholesterol (Fig. 5c) or of neutral POPC vesicles (Fig. 5d). For completeness, the difference between the binding of F17W and F11W was also calculated (Fig. 5a–d, open diamonds). The differences are smaller and may also be explained by experimental uncertainties.
We also tested whether the additional proline residue at position 0 had an effect on the membrane association of synthetic M1W peptides. The membrane-association constants for PWATLEKLMKAFESLKSF and WATLEKLMKAFESLKSF agreed well with those obtained from the recombinantly expressed products (not shown).
Collisional acrylamide quenching and REES
After having tested that tryptophan replacements and the additional proline do not significantly affect the secondary structure, alignment of the helical domain relative to the membrane normal (Michalek et al. 2013a), or the lipid-association, solvent accessibility and membrane penetration depth of the tryptophans was analyzed, for the first time, by use of fluorescence spectroscopy. Initially, acrylamide was used to analyze the collisional quenching of embedded tryptophan residues interacting with lipid vesicles of different lipid composition. The rate of quenching provides information about the accessibility of the tryptophans to the aqueous environment. Figure 3 depicts the Stern–Vollmer plots and fitted curves for M1W, F11W, and F17W peptides in buffer and in presence of 1.8 mM lipid LUVs (Fig. 3a–c). Unbound peptides in buffer undergo greater quenching than lipid-bound peptides, indicating that insertion of the tryptophan residue into the hydrophobic microenvironment results in its becoming inaccessible to the quenching agent. The corresponding Stern–Vollmer quenching constants are summarized in Table 2.

Collisional quenching of tryptophan fluorescence emission and REES for Htt17-W derivatives in the presence of LUVs of different lipid composition. The Stern–Vollmer plots for collisional quenching by membrane-impermeable acrylamide are depicted for a M1W, b F11W, and c F17 W in buffer (filled diamonds) and in the presence of POPC–POPS 75:25 mol/mol (filled circles), POPC–POPS–cholesterol 45:15:25 mol/mol/mol (open squares), POPE–POPG 75:25 mol/mol (open triangles), and pure POPC (filled inverted triangles) at a P/L ratio of 56. For calculating Stern–Vollmer quenching constants, curve fitting was performed as described in the “Materials and methods” section. Red edge excitation shift measurements (REES) were made for d M1W, e F11W, and f F17W under conditions similar to those used for a–c by varying the excitation wavelength from 280 to 315 nm. Data points were fitted sigmoidally
Quenching is least efficient for peptides associated with POPC–POPS vesicles. The quenching constant decreased by a factor of 4.5–11, which was in a range similar to that for tryptophan residues bound to POPE–POPG vesicles. For binding to POPC–POPS vesicles containing cholesterol, the Stern–Vollmer constants increased slightly, and interaction with zwitterionic POPC vesicles resulted in the highest quenching constant for the lipid mixtures. This was indicative of less shielded tryptophan residues caused by a binding equilibrium favoring the unbound peptide (Table 2).
REES was used to analyze the microenvironment of tryptophan residues in aqueous or hydrophobic environments. REES measurements give information about the rate of relaxation of the tryptophan’s solvent environment (Chattopadhyay et al. 2003) and are shown in Fig. 3 for M1W, F11W, and F17W under aqueous or lipid conditions. The different fluorescence maximum wavelength shifts to the red edge of the absorption band obtained by varying the excitation wavelength from 280 to 310 nm in buffer or in the presence of 1.8 mM lipid are summarized in Table 2. Pronounced shifts of 4 and 8 nm in buffer and 31 and 15 nm in the presence of POPC–POPS vesicles were observed for the peptides F11W and F17W, respectively (Fig. 3d, e). Incorporation of cholesterol reduced these shifts to 13 nm for F11W, whereas that for F17W was not affected. The smallest red shifts of 6 and 13 nm were observed in the presence of POPC vesicles; this was indicative of a solvent environment with high motional relaxation close to that of bulk water. These experiments therefore further support the binding and quenching experiments and are congruous with a different average location of the tryptophan residues as a function of lipid composition (Chattopadhyay et al. 2003).
The REES shift for M1W in buffer is greater than in the presence of lipid vesicles. Although the trend of shift differences for each lipid mixture is still the same as for F11W and F17W, the greater shift of 16 nm in buffer reflects slower relaxation of the environmental solvent (e.g. bulk water). This can be explained by hydrophobic interactions of the tryptophan indole ring with the benzyl rings of phenylalanines 11 and 17 in solution. Stacking caused by cation-π interaction induces non-exposed conformations of the tryptophan inside a more hydrophobic environment compared with the lipid-bound conformation on the lipid bilayer (or bilayer interface). This is compatible with size-exclusion measurements (Fig. 1b); those for M1W were indicative of a shorter retention time than those for F11W, F17W, and Htt17-syn.
Insertion depth of Htt17-W derivatives
To analyze the depth of insertion of the Htt17 domain, the M1W, F11W, and F17W peptides were successively incubated with LUVs containing 30 mol % POPC dibrominated at acyl-chain positions 6 and 7, 9 and 10, and 11 and 12, respectively. Previous experiments had revealed high lipid-binding affinity, efficient protection against the acrylamide quencher, and the largest REES shifts in the presence of POPC–POPS vesicles; this lipid composition was therefore used for examination of the maximum penetration of the three peptides. Quenching of the fluorescence intensity was monitored in the presence of 0.9 mM total lipid (Fig. 4a); the results obtained by distribution analysis of the data (Ladokhin et al. 1993) are given in Fig. 4b. The calculated average distance from the center of the lipid bilayer for the terminal tryptophan positions M1W and F17W were 11.2 ± 1.4 and 10.8 ± 1.2 Å, respectively, indicating interfacial location of the termini of Htt17. In contrast, F11W penetrated significantly more deeply, 7.3 ± 1.3 Å, into POPC–POPS vesicles, indicating location in the acyl-chain region of the lipid bilayer (Chattopadhyay and London 1987). Taken together, these results are indicative of interfacial location of the huntingtin 1–17 domain together with hydrophobic anchoring positioning of residue 11.

Depth-dependent quenching of Htt17-W derivatives by LUVs consisting brominated POPC lipids. a Corresponding changes in the emission fluorescence intensity are shown for M1W, F11W, and F17W peptides in presence of POPC–POPS LUVs consisting 30 mol % brominated POPC lipids in a molar P/L ratio of 56. The results are means from triplicate experiments, with error bars indicating the standard deviation. b Distribution analysis revealed the penetration depths of the substituted Htt17 peptides inside the lipid membranes
Discussion
The amino-terminus of the huntingtin protein, Htt17, is of crucial importance to the cellular location of the protein in the endoplasmic reticulum, Golgi apparatus, and mitochondria (Rockabrand et al. 2007; Atwal et al. 2007). Quantification of lipid–peptide interactions for chemically synthesized Htt17 peptides revealed conformational transitions from random coil to α-helix on interaction with lipid membranes of different composition (Michalek et al. 2013a). Until now, the effect of the very first, unstructured amino-acids during the lipid-binding interaction and details of the location, for example penetration depth of the peptide as a whole relative to the bilayer surface, had not been investigated.
Here, we used the tryptophan-substituted Htt17 derivatives M1W, F11W, and F17W to deepen our understanding of the functions of these positions in the lipid-binding process. These substitutions are located on three different structural elements of the Htt17 peptide: M1W in the amino-terminal region, which has a random coil secondary structure, as derived from NMR structural investigations, F11W in the center, and F17W at the very end of the amphipathic α-helix (Michalek et al. 2013b). Molecular size-exclusion chromatography and CD spectroscopy revealed compatible non-aggregated monomeric states in solution and α-helical content on interaction with the lipid bilayer. These findings are in agreement with previous results for the unmodified Htt17 peptide (Michalek et al. 2013a). Because tryptophan substitution altered the mean hydrophobicity to a minor extent only, membrane affinities were in the same range (compare Table 1 and Michalek et al. (2013a)), helix content was unaffected by tryptophan substitution (Fig. 1 and Michalek et al. 2013a)), and alignment of the helical domain was independent of proline addition (Michalek et al. 2013a), these results provide structural and lipid-dependent insight akin to that obtained for the natural Htt17 peptide.
To determine apparent dissociation constants of the termini and a central position of Htt17, intrinsic tryptophan emission fluorescence measurements were performed as a function of membrane lipid composition. The lipid interactions with POPC–POPS 3:1, POPE–POPG 3:1, POPC–POPS–cholesterol 3:1:2.7, and pure POPC were tested, thereby reflecting model membranes of such cellular compartments as the cytoplasmic membrane, mitochondria, and ER. All three Htt17-W derivatives had apparent dissociation constants similar to those of other lipid-interacting peptides, e.g. antimicrobial peptides (Sood et al. 2007; Silvestro et al. 1997; Vogt and Bechinger 1999; Christiaens et al. 2002). The K d values were in the order POPC–POPS < POPE–POPG < POPC–POPS–chol < POPC, and consistent in magnitude with the binding affinity of the unmodified Htt17 peptide, previously determined by CD spectroscopy (Michalek et al. 2013a). Moreover, the peptide with carboxy-terminal substitution (F17W) had a substantially higher K d value on interaction with POPE–POPG than on interaction with POPC–POPS, suggesting that details such as hydrogen bonding capacity, specific interactions with one lipid, membrane insertion depth, and the resulting curvature strain are important properties quantitatively explaining membrane binding (Bechinger et al. 1999). Moreover, on addition of cholesterol to lipid vesicles, the fluorescence intensity decreased. In several previous studies with antimicrobial lipid permeabilizing peptides, for example temporin, nisin, or LL37 similar, observations were made. Oligomerization of peptides, especially in the presence of cholesterol, reasonably explain these data (Chattopadhyay et al. 2003; Pina et al. 2007; Huster and Gawrisch 1999).
Whereas K d values were previously determined by following overall changes of secondary structure on addition of lipid (Michalek et al. 2013a) here we monitored changes in the fluorescence signals from different positions within the peptide (Aisenbrey et al. 2008). Most strikingly, for a given membrane composition the membrane dissociation constants varied substantially when the different tryptophan positions were compared with each other (Table 2). This could be because of changes in the peptide–membrane interactions as a result of the tryptophan replacements. However, such alterations do not seem to have profound consequences, because the overall topology of the helical domains of the tryptophan mutants (Fig. 5) agree well with the membrane alignment determined by a combination of solid-state and solution NMR spectroscopy of the native or P-Htt17 peptides (Michalek et al. 2013a, b). Therefore, we believe the polypeptide associated with the membrane through intermediate states, which would also explain the multi-phase behavior observed during the titration when the fluorescence intensities are monitored (Fig. 2d–f). The membrane-association constants suggest that the amino terminus interacts with the membrane before the helical domain encompassing residues 11 and 17 becomes inserted into the lipid bilayer. To analyze this scenario in more detail, the fractions of the peptide for which residue 1 only, residues 11 and 17, or all three sites resulted in changes in fluorescence emission wavelength when in a more hydrophobic environment are plotted in Fig. 5. These results suggest that at the onset of lipid titration approximately half the peptides bind with the amino terminus only, whereas complete membrane insertion concomitant with binding of additional peptide occurs when more lipids are added and the membrane surface correspondingly expands (Fig. 5e). Notably, the membrane association is identical within experimental error in the absence or presence of the additional proline (not shown). At a concentration of 0.1 mM POPC–POPS (LUVs) approximately 60 % of the peptide is membrane-associated, which is equivalent to one peptide per 5 or 6 lipid molecules (only one monolayer of which may be accessible). Steric crowding at such high peptide densities could indeed explain why full helix insertion is delayed (Aisenbrey et al. 2008). Such a dense crowding of the membrane surface also agrees with the observation of fluorescence quenching during the initial steps of the titration (Fig. 2d–f). Whereas fluorescence intensity increases when more lipid is added, a decrease is again observable in the final steps of the titration. These results are indicative of profound structural rearrangement during the titration process and is suggestive of oligomerization of the peptide. The formation of dimers was indeed suggested to explain the somewhat unexpected pitch angle of the amphipathic α-helix when investigated by solid-state NMR spectroscopy (Michalek et al. 2013a, b).

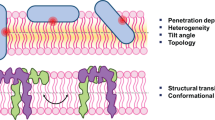
Membrane-association intermediates form as a function of lipid concentration. The results shown in Fig. 2 were used to calculate the fraction of peptide when residue 1 only (open circles), residues 1 and 17 (open diamonds), or all three residues (filled inverted triangles) are exposed to a hydrophobic environment. The results were normalized in such a manner that the amount of peptide with all three sites in a membrane environment at 1.8 mM lipid corresponds to unity, even though in several of the experiments a significant amount of peptide remained exposed to the water phase. The compositions of the LUV added were: a POPC–POPS 75:25, b POPC–POPS–cholesterol 45:15:25, c POPE–POPG 75:25, and d POPC. e The model shows the successive steps of membrane association. Peptide crowding appears, because of amino-terminal association of the peptide with the membrane. Membrane expansion facilitates interaction of the lipid with the carboxy-terminal part of the peptide. This results in deep penetration of residue 11 followed by conformational changes to α-helix. Finally, the α-helix anchors at the lipid interface with exposed termini in an in-plane orientation
Moreover, experiments with collisional quenching by acrylamide and REES were consistent with the investigation of the Htt17-W derivatives on lipid insertion. All three tryptophan positions inserted into the lipid bilayer, accompanied with low Stern–Vollmer quenching constants and high REES shift compared with peptides in buffer. Again, the quenching experiments and REES were in good agreement with tryptophan insertion measurements and independently strengthened the order of lipid specificity already described. However, unusually high REES shifts were observed for M1W in buffer, consistent with stacking of aromatic side chains induced by hydrophobic and π-interactions, which exclude water molecules surrounding the indole ring of the tryptophan residue. A similar observation has been made for the shiga-toxin B subunit of Shigella dysenteriae (Pina et al. 2007). In this latter case the stacking finally leads to a less extended random coil conformation, which was revealed by molecular size-exclusion chromatography. On lipid binding, the tryptophan residue has an exposed conformation surrounded by bulk water and water associated with the bilayer interfacial region, accompanied by high rates of relaxation of the solvent, causing smaller REES shifts.
Investigation of the depth of penetration of Htt17-W derivatives by brominated POPC lipids revealed deeper insertion of the center tryptophan F11W compared with terminal tryptophan substitutions inside POPC–POPS vesicles. In previous studies, the disorder of lipid membranes has been shown by 1H-PFG-MAS NOESY spectroscopy, in which magnetization transfer between terminal methyl groups of the lipid acyl chain and choline protons has been observed (Huster and Gawrisch 1999). These choline-to-chain cross peaks have also been shown for POPC membranes as a result of occasional penetration of water into the hydrophobic core (Gawrisch et al. 2007). This means that exact estimation of peptide location by use of brominated lipids is also affected by membrane disordering events and, here, enables determination of the relative positioning of the tryptophans only.
Previous studies using oriented solid-state NMR measurements have revealed orientation of the α-helical backbone of Htt17 in plane to the membrane normal, with pitch and tilt angles of 137° and 103°, respectively (Michalek et al. 2013a). The insertion difference of 3–4 Å when F11W is compared with M1W and F17W reveals its isolated location in the helical wheel (Fig. 1a) and possibly reflects different rotameric states of the tryptophan side chain along the α-helical axis. At the carboxy and amino-termini Cα–Cβ (χ 1 torsion angle) and Cβ–Cγ (χ 2 torsion angle) rotation is less restricted than in the center of the α-helix, where the side chain adopts a more extended conformation (Clayton and Sawyer 1999). This explanation is supported by the observed Stern–Vollmer quenching constants and the REES shift for F11W, which are the lowest and the highest of all measurements made, respectively, thereby reflecting the inherent importance of phenylalanine 11 in the lipid-anchoring process.
Taken these experiments together, the model for the lipid binding properties of Htt17, summarized in Fig. 5e could be expanded. Notably the Htt17 domain has all the functional characteristics of a lipid anchor for the huntingtin protein, and the deep insertion of phenylalanine 11 strongly stabilizes the lipid-bound polypeptide.
Conclusion
In summary, our results derived from independent measurement of tryptophan emission fluorescence strengthen our understanding of how the Htt17 domain is anchored at lipid membranes in an in-plane orientation. Here, we determined, for the first time, the depth of penetration of the peptide, which brings residue 11 of the helical domain to approximately 7 Å from the membrane center. This reveals the importance of this aromatic side-chain position to lipid-anchoring properties when at the same time the bulk of the Htt17 peptide is located at the interfacial region of the phospholipid head groups. The amino terminus of P-Htt17 participates in peptide–lipid interactions and establishes the first contacts during lipid titration. The carboxy-terminal residue remains mostly exposed to the aqueous environment, thereby facilitating interaction between the neighboring polyglutamine of the huntingtin protein and the biological lipid bilayer. Notably, the fluorescence quenching observed even at low peptide-to-lipid ratios provides additional evidence of the formation of oligomeric structures in membrane environments.
Abbreviations
- CD:
-
Circular dichroism
- DPC:
-
Dodecylphosphocholine
- HFIP:
-
1,1,1,3,3,3-Hexafluoro-2-propanol
- Htt17:
-
Huntingtin 1–17
- NMR:
-
Nuclear magnetic resonance
- POPC:
-
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPE:
-
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPG:
-
1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho(1′-rac-glycerol)
- POPS:
-
1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- Chol:
-
Cholesterol
- TFA:
-
Trifluoroacetic acid
- REES:
-
Red-edge excitation shift
References
Aisenbrey C, Bechinger B, Grobner G (2008) Macromolecular crowding at membrane interfaces: adsorption and alignment of membrane peptides. J Mol Biol 375:376–385
Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R (2007) Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet 16:2600–2615
Bechinger B, Kinder R, Helmle M, Vogt TC, Harzer U, Schinzel S (1999) Peptide structural analysis by solid-state NMR spectroscopy. Biopolymers 51:174–190
Bechinger B, Aisenbrey C, Bertani P (2004) The alignment, structure and dynamics of membrane-associated polypeptides by solid-state NMR spectroscopy. Biochim Biophys Acta 1666:190–204
Bolen EJ, Holloway PW (1990) Quenching of tryptophan fluorescence by brominated phospholipid. Biochemistry 29:9638–9643
Chang YC, Ludescher RD (1994) Local conformation of rabbit skeletal myosin rod filaments probed by intrinsic tryptophan fluorescence. Biochemistry 33:2313–2321
Chattopadhyay A, London E (1987) Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 26:39–45
Chattopadhyay A, Rawat SS, Kelkar DA, Ray S, Chakrabarti A (2003) Organization and dynamics of tryptophan residues in erythroid spectrin: novel structural features of denatured spectrin revealed by the wavelength-selective fluorescence approach. Protein Sci 12:2389–2403
Chen S, Wetzel R (2001) Solubilization and disaggregation of polyglutamine peptides. Protein Sci 10:887–891
Christiaens B, Symoens S, Verheyden S, Engelborghs Y, Joliot A, Prochiantz A, Vandekerckhove J, Rosseneu M, Vanloo B (2002) Tryptophan fluorescence study of the interaction of penetratin peptides with model membranes. Eur J Biochem 269:2918–2926
Clayton AH, Sawyer WH (1999) Tryptophan rotamer distributions in amphipathic peptides at a lipid surface. Biophys J 76:3235–3242
Demchenko AP, Ladokhin AS (1988) Red-edge-excitation fluorescence spectroscopy of indole and tryptophan. Eur Biophys J 15:369–379
Dubreil L, Compoint JP, Marion D (1997) Interaction of puroindolines with wheat flour polar lipids determines their foaming properties. J Agric Food Chem 45:108–116
Eisenberg D, Schwarz E, Komaromy M, Wall R (1984) Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol 179:125–142
Gawrisch K, Gaede HC, Mihailescu M, White SH (2007) Hydration of POPC bilayers studied by 1H-PFG-MAS-NOESY and neutron diffraction. Eur Biophys J 36:281–291
Ghosh AK, Rukmini R, Chattopadhyay A (1997) Modulation of tryptophan environment in membrane-bound melittin by negatively charged phospholipids: implications in membrane organization and function. Biochemistry 36:14291–14305
Hill AV (1910) The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J Physiol 40:4–7
Huster D, Gawrisch K (1999) NOESY NMR crosspeaks between lipid headgroups and hydrocarbon chains: spin diffusion or molecular disorder? J Am Chem Soc 121:1992–1993
Kegel KB, Sapp E, Yoder J, Cuiffo B, Sobin L, Kim YJ, Qin ZH, Hayden MR, Aronin N, Scott DL, Isenberg G, Goldmann WH, DiFiglia M (2005) Huntingtin associates with acidic phospholipids at the plasma membrane. J Biol Chem 280:36464–36473
Ladokhin AS, Wang L, Steggles AW, Malak H, Holloway PW (1993) Fluorescence study of a temperature-induced conversion from the “loose” to the “tight” binding form of membrane-bound cytochrome b5. Biochemistry 32:6951–6956
Macdonald PM, Seelig J (1988) Anion binding to neutral and positively charged lipid membranes. Biochemistry 27:6769–6775
McIntosh TJ, Holloway PW (1987) Determination of the depth of bromine atoms in bilayers formed from bromolipid probes. Biochemistry 26:1783–1788
Michalek M, Salnikov ES, Werten S, Bechinger B (2013a) Membrane interactions of the amphipathic amino terminus of huntingtin. Biochemistry 52:847–858
Michalek M, Salnikov ES, Bechinger B (2013b) Structure and topology of the huntingtin 1–17 membrane anchor by a combined solution and solid-state NMR approach. Biophys J 105:699–710
Mosior M, McLaughlin S (1991) Peptides that mimic the pseudosubstrate region of protein kinase C bind to acidic lipids in membranes. Biophys J 60:149–159
Mosior M, McLaughlin S (1992) Binding of basic peptides to acidic lipids in membranes: effects of inserting alanine(s) between the basic residues. Biochemistry 31:1767–1773
Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ (2008) N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci 28:2783–2792
Pan CP, Callis PR, Barkley MD (2006) Dependence of tryptophan emission wavelength on conformation in cyclic hexapeptides. J Phys Chem B 110:7009–7016
Pina DG, Johannes L, Castanho MA (2007) Shiga toxin B-subunit sequential binding to its natural receptor in lipid membranes. Biochim Biophys Acta 1768:628–636
Rhoades E, Ramlall TF, Webb WW, Eliezer D (2006) Quantification of alpha-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys J 90:4692–4700
Rockabrand E, Slepko N, Pantalone A, Nukala VN, Kazantsev A, Marsh JL, Sullivan PG, Steffan JS, Sensi SL, Thompson LM (2007) The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet 16:61–77
Silvestro L, Gupta K, Weiser JN, Axelsen PH (1997) The concentration-dependent membrane activity of cecropin A. Biochemistry 36:11452–11460
Sood R, Domanov Y, Kinnunen PK (2007) Fluorescent temporin B derivative and its binding to liposomes. J Fluoresc 17:223–234
Sreerama N, Woody RW (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem 287:252–260
Tam S, Geller R, Spiess C, Frydman J (2006) The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol 8:1155–1162
Tam S, Spiess C, Auyeung W, Joachimiak L, Chen B, Poirier MA, Frydman J (2009) The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat Struct Mol Biol 16:1279–1285
Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJ, Anjum DH, Kodali R, Creamer TP, Conway JF, Gronenborn AM, Wetzel R (2009) Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol 16:380–389
Vogt TC, Bechinger B (1999) The interactions of histidine-containing amphipathic helical peptide antibiotics with lipid bilayers. The effects of charges and pH. J Biol Chem 274:29115–29121
Wieprecht T, Apostolov O, Beyermann M, Seelig J (2000) Membrane binding and pore formation of the antibacterial peptide PGLa: thermodynamic and mechanistic aspects. Biochemistry 39:442–452
Acknowledgments
The authors gratefully acknowledge financial support by the CHDI foundation (early discovery initiative) and the Deutsche Forschungsgemeinschaft (DFG), for supporting MM by contributing to editing of the manuscript. We are indebted to the Agence Nationale de la Recherche (projects LabEx Chemistry of Complex Systems and Prolipin), the RTRA International Center of Frontier Research in Chemistry, the University of Strasbourg, the CNRS, and the Région Alsace. We are, moreover, grateful to Delphine Hatey for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Michalek, M., Aisenbrey, C. & Bechinger, B. Investigation of membrane penetration depth and interactions of the amino-terminal domain of huntingtin: refined analysis by tryptophan fluorescence measurement. Eur Biophys J 43, 347–360 (2014). https://doi.org/10.1007/s00249-014-0966-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-014-0966-9