
Abstract
Both the structure and function of host-associated microbial communities are potentially impacted by environmental conditions, just as the outcomes of many free-living species interactions are context-dependent. Many amphibian populations have declined around the globe due to the fungal skin pathogen, Batrachochytrium dendrobatidis (Bd), but enivronmental conditions may influence disease dynamics. For instance, in Panamá, the most severe Bd outbreaks have occurred at high elevation sites. Some amphibian species harbor bacterial skin communities that can inhibit the growth of Bd, and therefore, there is interest in understanding whether environmental context could also alter these host-associated microbial communities in a way that might ultimately impact Bd dynamics. In a field survey in Panamá, we assessed skin bacterial communities (16S rRNA amplicon sequencing) and metabolite profiles (HPLC-UV/Vis) of Silverstoneia flotator from three high- and three low-elevation populations representing a range of environmental conditions. Across elevations, frogs had similar skin bacterial communities, although one lowland site appeared to differ. Interestingly, we found that bacterial richness decreased from west to east, coinciding with the direction of Bd spread through Panamá. Moreover, metabolite profiles suggested potential functional variation among frog populations and between elevations. While the frogs have similar bacterial community structure, the local environment might shape the metabolite profiles. Ultimately, host-associated community structure and function could be dependent on environmental conditions, which could ultimately influence host disease susceptibility across sites.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Host-associated bacteria play key roles in development, reproduction, digestion, and protection against pathogens for their hosts [1]. In recent decades, culture-independent molecular techniques have rapidly expanded our knowledge of the diversity, ubiquity, and functional ability of these symbiotic communities. In addition, the integration of different molecular techniques and “omics” methods, such as metabolomics combined with 16S ribosomal RNA (rRNA) gene amplicon sequencing, is providing an opportunity to address important ecological questions, including insights into the potential link between community structure and function in complex microbial communities [2, 3]. For example, the use of 16S rRNA gene amplicon sequencing has provided important insights into the structure-function relationship in soil microbial communities, where functional redundancy might be present in important soil microbial processes [4], and in the gut microbiome of mammals, in which functional repertoires of microbiome genes can be predicted by the structure of the bacterial community [5].
One key function of host-associated microbes on animal hosts is host defense against pathogens, which has been observed across distantly related host taxa, including corals, shrimp, Hydra, insects, birds, humans, and amphibians [6–11]. For example, amphibians harbor diverse skin bacterial communities that appear to be host species-specific [12–15]. Some of these skin bacteria can protect their amphibian hosts from the pathogenic fungus Batrachochytrium dendrobatidis (Bd) [15–19] seemingly through the production of secondary metabolites that inhibit Bd growth [18, 20, 21]. As Bd has been associated with drastic global amphibian population declines [22] and infects over 500 amphibian species across the globe [23], interest has grown in understanding the factors that shape these microbial communities and their function.
In general, relatively little is known about how environmental factors influence the structure and function of host-associated microbial communities, although there is certainly appreciation more broadly for the role of context dependency in determining the outcome of symbiotic interactions [24–26]. For example, for host-pathogen interactions, changes in temperature can alter transmission rates of pathogenic fungi from adult bark beetles to pine trees [27], enhance bleaching in corals [28, 29], and influence the morbidity of white-nose syndrome in bats [30]. Chytridiomycosis outbreaks in amphibians, caused by Bd infections, have varied in intensity across space and time, which suggests a context dependency in the outcome of interactions among the amphibian host, Bd, and the environment [31, 32]. In the Neotropics, amphibian diversity losses due to Bd have been substantial [33], but in general, Bd-induced population declines and local extinctions have been more dramatic at higher elevations relative to the lowlands [34–37], potentially because of variation in environmental factors (e.g., temperature and rainfall) that alter disease outcomes.
Indeed, context-dependent outcomes of host-microbiome-pathogen interactions could play a critical role in wildlife diseases [32]. But while some effort has focused on understanding how amphibian-Bd interactions might change across elevations (e.g., [38–42]), little work has addressed changes in the amphibian-microbiome interaction with elevation. In the present study, we aimed to determine how the diversity, structure, and potential function (represented by metabolite profiles) of skin bacterial communities on amphibians vary with elevation. Specifically, we sampled populations of the species Silverstoneia flotator—a species thought to be tolerant to Bd and with a broad distributional range at both highland and lowland sites. We hypothesized that we would observe distinct bacterial communities and metabolite profiles across elevations given that abiotic factors, such as precipitation, temperature ranges, and strength of seasonality, vary along elevational gradients. Importantly, these environmental factors could directly or indirectly influence the skin bacterial communities.
Material and Methods
Silverstoneia flotator (Family: Dendrobatidae, Dunn 1931) occurs from “5 to 1050 m” in Panamá [43]. Across this elevational range, vegetation changes from lowland wet/moist forest (<600 m) to premontane forest (600–1500 m), which is associated with changes in climatic factors such as temperature and precipitation [43]. S. flotator is a common terrestrial diurnal species occurring in leaf litter, including along streams. This species is broadly distributed across Panamá and may be tolerant of Bd infection considering it has not declined following Bd arrival at one highland site [33] and that there are populations persisting with Bd at other highland sites [36, 42].
Sample Collection
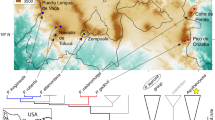
In 2013, we sampled 10–15 S. flotator at each of six sites where Bd is known to occur to assess their skin bacterial communities, skin metabolite profiles, and Bd infection status. Of the six sites, three were highland sites above 600 m, while the other three were lowland sites below 300 m (Table 1; Fig. 1). Among the study sites, chytridiomycosis has caused amphibian declines at two of the highland sites, El Cope and Cerro La Campana [34, 36], while there have been no declines at one of the lowland sites, Soberanía, despite the presence of Bd [36]. There are no published studies for the other three sites describing the impact of Bd arrival on the amphibian community, though it is known that Bd is present at these sites (R. Ibáñez and J. Voyles pers. comm.).

Map of Panamá showing the location of field sites throughout the country
During sampling, we handled each frog with a new pair of gloves and placed it in a sterile Whirl-Pak® bag (Nasco, Fort Atkinson, WI, USA) until being swabbed within 30 min. The swabbing protocol was conducted as in Belden et al. [14]; prior to swabbing, each frog was rinsed with 50 mL of sterile deionized water to remove any dirt and transient bacteria [12, 14, 44]. Then, each frog was swabbed twice: the first swab was used to assess the skin bacterial community and Bd infection status, and the second swab was used to assess the metabolite profile. The first swab was a sterile rayon swab (MW113, Medical Wire Equipment), and the second swab was a pre-treated polyurethane swab (14-960-3J, Fisher Scientific). The polyurethane swabs were pre-treated to remove methanol-soluble impurities by stirring them in a methanol wash and letting them be air-dried in a fume hood [45]. Each swab collection consisted of 20 strokes on the ventral surface and 5 strokes in a single direction on each thigh and hind foot for a total of 40 strokes per swab. The swabs were individually placed in 1.5-mL centrifuge tubes and stored in a cooler either with dry ice or with ice packs (samples from Soberanía, Cerro Azul, and Sierra Llorona) or in liquid nitrogen (Cope, LowCope, and Campana) during fieldwork. Temperature variation in short-term sample storage does not influence the assessment of bacterial community composition [46]. In the laboratory, swabs were placed at −80 °C until processing. Frogs were returned to the site of capture after swabbing.
Data from 60 frogs (N = 5–12/site) was used for the comparative analyses of the skin bacterial communities (Table 1). We expected to find a low incidence of Bd infection on S. flotator since it persists in sites with Bd and because we sampled during the beginning of the rainy season, which could be a time of low seasonal prevalence of infection in the Neotropics (e.g., data from Costa Rica [47]). Of the 60 frogs we swabbed, we preliminarily selected 40 individuals from four sites (two highlands: Cope High and Cerro Azul; and two lowlands: LowCope and Soberanía) to assess Bd infection status. For the metabolite profiles, we included a total of 72 individuals from five sites (N = 12–15/site, with no metabolite swabs from Sierra Llorona; Table 1). Out of these 72 individuals, 55 overlapped with the set of 60 used for the bacterial community analyses.
Sample and Data Processing
Skin Bacterial Communities
DNA was extracted from the first swab using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol and with an initial lysozyme incubation at 37 °C for 1 h. A final volume of 100 μL of DNA was obtained, which was used as the template DNA for the Bd infection analyses as well.
Skin bacterial communities were assessed by amplifying and sequencing the V4 region of the 16S rRNA gene with the primers 515F and 806R [48]. The barcoded reverse primer contained a 12-base error-correcting Golay code to tag PCR products, which allowed for multiplexing. PCR reactions (25 μL) were prepared with 12 μL of molecular-grade PCR water, 10 μL of 5 Prime Hot Master Mix, 0.5 μL of each primer (10 μM concentration), and 2.0 μL of template DNA. PCRs were run in triplicate with a control that did not contain template DNA. PCR reactions were conducted with an initial cycle of 3 min at 94 °C, followed by 35 cycles of 45 s at 94 °C, 60 s at 50 °C, and 90 s at 72 °C, with a final extension of 10 min at 72 °C. PCR products from triplicate reactions were pooled and visualized on a 1% agarose gel and quantified using a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, California). PCR products of each sample (200 ng/sample; N = 60) were pooled to make a composite sample and cleaned with the QIAquick PCR Purification Kit (Qiagen, Valencia, CA). The final pooled sample was sent for sequencing on an Illumina MiSeq instrument with a 250-bp paired-end strategy at the Dana-Farber Cancer Institute of Harvard University following Caporaso et al. [48].
Raw sequences from the Illumina 16S rRNA gene amplicon sequencing were assembled with Fastq-Join [49]. Assembled sequences were processed and quality-filtered using the default parameters of the Quantitative Insights Into Microbial Ecology (QIIME) pipeline [50], with the exception that we allowed for no errors in the barcodes, increased the number of minimum consecutive low-quality base calls allowed before truncating a read (r) to 10, and decreased the fraction of the minimum number of consecutive high-quality base calls to include a read (p) to 0.5. Sequences were clustered into operational taxonomic units (OTUs, ~bacterial species) based on a 97% similarity threshold using the UCLUST method [51], and OTUs were represented by the most abundant sequence. The representative sequences were aligned to the Greengenes 13_5 reference database [52] using PyNAST [53] and assigned taxonomy using the RDP classifier [54]. We removed OTUs with fewer than 0.001% of the total number of sequences [55]. The sequencing depth per sample ranged from 6132 to 96,126, so we rarefied the dataset at a depth of 6000 reads/sample. The final dataset consisted of 3497 OTUs across the 60 samples, with 193–1079 OTUs (mean = 491 ± 186) per frog.
Skin Metabolite Profiles
To assess the metabolite profiles, we isolated and characterized small molecules from the swabs using HPLC-UV/Vis detection [45]. This method is useful for identifying metabolite patterns across environmental gradients, even though we cannot identify individual compounds. Given the nature of the method, highly polar compounds, such as peptides and proteins that might be produced by the amphibian host, are not detected; thus, the compounds detected represented small molecules hypothesized to be bacterial secondary metabolites [45]. For the characterization of the metabolite profiles, swabs were sent frozen to Villanova University and processed as detailed in Belden et al. [14].
Compounds eluted from samples were characterized by retention times and UV-Vis chromophore. Retention times of all detected compounds (peaks) were normalized to that of a naphthalene internal standard (20.69 min). The retention time of each chromatographic feature was determined manually using Applied Biosystems Analyst software V.1.5.1. The generated dataset was then revised to account for slight variations in retention time across multiple samples, with a focus on major chemical components. Compounds present in multiple samples that displayed both similar retention times and identical spectroscopic features were assigned as a single compound, and unique compounds only detected in a single sample were considered noise and therefore disregarded. Lastly, chromatographic features with less than 3000 mAU × min were considered minor components and disregarded.
Bd Detection and Quantification
The 40 samples assessed for Bd were analyzed with a TaqMan real-time PCR (qPCR) assay developed by Boyle et al. [56]. We amplified ITS-1-5.8S of the Bd genome using primers ITS1–3 Chytr and 5.8S Chytr and the probe MGB2. DNA standards were prepared by making serial dilutions for 1000–0.1-zoospore genomic equivalents of the strain JEL 310, a strain isolated from Smilisca phaeota from Fortuna, Panamá [57]. Given that the copy number of the region ITS-1-5.8S of the strain used for the standards is unknown, a low value equal to one copy number could not be estimated, as described by Rebollar et al. [37]. Thus, a sample was considered infected when the values estimated by the qPCR assay were above 0.1-zoospore genomic equivalents. The samples were run in duplicate and considered positive when amplification was observed in both replicates. The infection intensity (i.e., number of zoospore equivalents) of a sample represents the average of both replicates.
Data Analysis and Statistical Methods
Overview
We assessed whether there were differences in skin bacterial community structure and metabolite profiles between lowland and highland populations of S. flotator, as well as structure-function relationships. As there was very little Bd infection detected, we were not able to assess variation based on Bd infection intensity or prevalence. We also described the core microbiome to provide information about the bacterial taxa associated with a neotropical species that has survived chytridiomycosis epidemics and persists at sites with endemic Bd. Unless noted, all analyses were conducted in R version 3.2.2 [58].
Alpha and Beta Diversity Analyses
Alpha diversity comparisons of skin bacterial communities between elevations and across sites were conducted for richness (OTUs/individual), phylogenetic diversity, and the Shannon index (H′, which assessed community evenness). These metrics were computed for each frog with QIIME. We compared alpha diversity estimates between elevations by fitting the metrics to generalized linear mixed models (GLMMs), in which the variable “Site” was included in the models as a random effect to account for the nestedness of individual frogs within each site. Among-site comparisons were performed by fitting the metrics to generalized linear models (GLMs). For richness estimates, we used a negative binomial error distribution with the log link function to account for overdispersion; for elevation comparisons, we used the function glmer.nb from the package lmer4 [59], and for among-site comparisons, we used the function glm.nb from the package MASS [60]. For Shannon and phylogenetic diversity estimates, we used a Gamma error distribution with the inverse link function to account for heteroskedasticity; for elevation and among-site comparisons, we used the functions glmer and glm from the package lmer4 [59], respectively. Multiple comparisons were conducted with Tukey tests using the function glht of the package multcomp [61], which includes multiple comparisons for generalized linear models. Moreover, we also compared metabolite richness (metabolites/frog) between elevations and among sites by fitting the metric to a linear mixed model (function lmer) and a linear model (function lm), respectively (packages lmer4 and stats). Visual assessments of residual plots with model predictions corroborated that these approaches were appropriate.
We performed GLMs to assess the effect of longitude (converted to UTM) on the alpha diversity estimates. GLMs were performed as described above, with a negative binomial error distribution for richness estimates and a Gamma error distribution for Shannon and phylogenetic diversity. Visual assessments of residual plots with model predictions corroborated that these approaches were appropriate.
To examine whether elevation and site explained variation in the structure of the bacterial communities and metabolite profiles (beta diversity), we performed permutational multivariate analyses of variance, PERMANOVA [62], based on a Bray-Curtis dissimilarity matrix for the OTUs and on a Jaccard dissimilarity matrix for the metabolites. Jaccard dissimilarities, which only take into account presence-absence, were used for the metabolite data because the HPLC-UV/Vis output does not allow for the estimation of relative abundance. Results were visualized using non-metric multi-dimensional scaling (NMDS). The distance matrices were calculated with the function vegdist, and PERMANOVAS were performed using the function adonis in the vegan package [63].
Examining Links Between Bacterial Community Structure and Metabolite Profiles
To assess potential relationships between bacterial community structure and metabolite profiles, we conducted a Mantel test. The Mantel test requires that the matrices being tested have the same samples, so we removed the 12 frogs from the metabolite dataset that were not represented in the OTU dataset. This resulted in the same 55 samples in both data matrices. We conducted the Mantel test in two ways: separately by individual sites, and then with all the sites together. Mantel tests were performed with the function mantel in the vegan package [63].
Incidence of Bd Infections
Bd was only detected in one of the highland sites (Cope), and in only three frogs, so no comparisons regarding Bd across sites or elevation could be conducted. Bd prevalence and infection intensity at Cope (zoospore equivalents) were estimated using the Wilson interval and a bootstrap approach (BCa; 10,000 permutations), respectively, using R [37]. As only three of the 40 frogs analyzed were Bd-positive, we did not quantify Bd in the remaining 20 frogs from the other two sites.
Core Microbiome
The core microbiome is defined as “organisms common across microbiomes” [64] and may be fundamental to community function [64]. The core microbiome for S. flotator was determined using the compute_core_microbiome script in QIIME and was defined as OTUs present on ≥90% of all the frogs. The relative abundances of the core OTUs, across frogs, sites, and elevations were visualized via a heatmap, using the function heatmap.2 in the package gplots [65]. We examined whether the relative abundance of each core OTU significantly differed based on elevation and site with a Kruskal-Wallis test. We calculated the corrected p values for the multiple comparisons based on the Bonferroni procedure using the group_significance script in QIIME.
Results
Bacterial Community: Diversity and Structure
None of the three metrics of alpha diversity of bacterial communities differed between high and low elevations (richness: χ 2 = 0.06, p = 0.81; Shannon: χ 2 = 0.01, p = 0.92; phylogenetic diversity: χ 2 = 0.24, p = 0.62, Table S1a). However, we observed significant differences among sites for all the metrics (richness: deviance = 60.62, p < 0.001; Shannon: F = 10.36, p < 0.001; phylogenetic diversity: F = 5.2, p < 0.001). Alpha diversity pairwise comparisons among sites showed a potential west to east trend, especially for richness and phylogenetic diversity estimates, where the sites west of the Panamá Canal had significantly higher diversity estimates than those on the east side (Fig. 2; Table S1b). The generalized linear models used to assess this pattern suggested a significant decline towards the east in the three alpha diversity metrics (richness: log-scale slope = −3.88E−06, SE = 7.27E−07, p = <0.001; Shannon: inverse-log scale slope = 5.55E−07, SE = 1.07E−07, p = <0.001; phylogenetic diversity: inverse transformed slope = 7.66E−08, SE = 1.67E−08, p = <0.001; Table S2, Fig. S1).

Alpha diversity of bacterial OTUs by site (richness, phylogenetic diversity, Shannon index)
Bacterial community structure (beta-diversity) differed between elevations and among sites when taking into account all the sites (Fig. 3a, b. NMDS stress 0.16; by elevation: adonis pseudo-F = 5.83, p < 0.001, R 2 = 0.09; by site: adonis pseudo-F = 5.13, p < 0.001, R 2 = 0.32). However, the differences in community structure and the variation explained by the factors “elevation” and “site” were likely driven by the distinct communities at LowCope, as shown on the NMDS. When LowCope was removed from the analysis, there was not a significant difference based on elevation or site for the remaining sites (by elevation: adonis pseudo-F = 1.03, p = 0.34, R 2 = 0.021; by site: adonis pseudo-F = 1.53, p = 0.12, R 2 = 0.12).

Beta diversity comparisons of bacterial communities using non-metric multi-dimensional scaling (NMDS) ordinations based on Bray-Curtis dissimilarity distances between elevation categories (a) and among-study sites (b). Each point represents a single frog, and point shapes: triangles = high elevation sites; circles = low elevation sites. Axis labels (MDS) represent the multi-dimensional scaling
Metabolite Profiles: Diversity and Structure
A total of 84 unique metabolites were identified (range 25–38/frog; mean ± SD 33.38 ± 2.52). Metabolite richness differed significantly between elevations, although the means were very similar (χ 2 = 5.73, p = 0.017; mean ± SD: high 33 ± 2.72; low 34 ± 1.97). There was no difference in metabolite richness among sites (F = 1.48, p = 0.217; mean ± SD: Campana 33 ± 2.47; Cerro Azul 33 ± 3.19; Cope 33 ± 2.73; CopeLow 34 ± 1.90; Soberanía 34 ± 2.11). Metabolite profiles differed between elevations and across sites (Fig. 4a, b; NMDS stress 0.14; by elevation: adonis pseudo-F = 10.37, p < 0.001, R 2 = 0.13; by site: adonis pseudo-F = 15.53, p < 0.001, R 2 = 0.48); however, site explained more of the observed variation (PERMANOVA: site R 2 = 0.48; elevation R 2 = 0.13). As one site (i.e., Soberanía) appeared to potentially drive this difference based on the NMDS ordination (Fig. 4b), we conducted a further analysis excluding the site “Soberanía.” However, there was still significant variation in metabolite profiles among sites after excluding Soberanía (adonis pseudo-F = 4.42, p < 0.001, R 2 = 0.20, Table S3). All pairwise comparisons of metabolite profiles among sites were significantly different, except for LowCope-Campana (Table S3).

Beta diversity comparisons of metabolite profiles using non-metric multi-dimensional scaling (NMDS) ordinations based on Jaccard dissimilarity distances between elevation categories (a) and among-study sites (b). Each point represents a single frog, and point shapes: triangles = high elevation sites; circles = low elevation sites. Axis labels (MDS) represent the multi-dimensional scaling
Linking Bacterial Community Structure and Metabolite Profiles
Based on Mantel tests, we found no correlation between the structure of the bacterial communities at the sites and their respective metabolite profiles (Campana: Mantel r statistic = −0.12, p = 0.65; Cerro Azul 0.04, p = 0.34; Cope 0.20, p = 0.16; LowCope 0.03, p = 0.36; Soberanía 0.11, p = 0.31; all sites together 0.06, p = 0.16).
Bd Occurrence
Prevalence of infection by Bd at Cope was 30% (3/10 frogs; CI 95% 11–60%), and the mean infection load per frog was 250 (CI 95% 98–347) zoospore genomic equivalents. No Bd was detected at the other three sites we tested.
Core Microbiome
Despite a total of 3497 OTUs identified on S. flotator, only 25 OTUs were in the 90% core microbiome (from nine families in three phyla: Actinobacteria, Firmicutes, and Proteobacteria; Table S4). In addition, the most abundant core OTUs (relative abundance ≥1%) were in the families Cellulomanadaceae (mean relative abundance of family 15%), Comamonadaceae (1%), Enterobacteriaceae (1%), Pseudomonadaceae (4%), and Xanthomonadaceae (1%). There was no significant difference in the relative abundance of any core OTUs between elevations (Fig. 5), except for one Comamonadaceae (ID number 4456068) that had higher mean relative abundance (χ 2 = 12.62, p = 0.010) at high elevation (0.015 vs. 0.008%). However, there was significant variation across sites for ten core OTUs (Fig. 5). This variation mainly reflects the difference at LowCope; six of the ten core OTUs occurred at a lower relative abundance there, and two Enterobacteriaceae (ID numbers 2119418 and 814442) and one Pseudomonadaceae (269930) occurred at a higher relative abundance (Enterobacteriaceae ID 2119418: χ 2 = 21.68, p = 0.02; Enterobacteriaceae ID 814442: χ 2 = 21.52, p = 0.02; Pseudomonadaceae ID 269930: χ 2 = 23.19, p = 0.008).

Heatmap of the relative abundances of the OTUs considered members of the core microbiome across samples and sites. Each row in the heatmap represents a single frog sampled. The colors next to rows are associated with a specific site. Columns represent each core OTU. OTUs with an asterisk (*) represent OTUs with significantly different relative abundances (p < 0.05) across sites. Red asterisks represent OTUs with a significant higher relative abundance at the LowCope site (color figure online)
Discussion
In our study, we used replication of highland and lowland sites to try to understand the potential role of environmental variation in the structure and function of skin bacterial communities. However, we did not see key differences in bacterial communities on S. flotator between high and low elevations, either in terms of alpha diversity metrics or in terms of bacterial community structure. Skin metabolite profiles, which we used as an estimate of microbial community function, did differ between elevations. This suggests that environmental conditions at high and low elevation sites might not contribute to the establishment of completely different bacterial communities, but they could result in a change in function of the communities. While we cannot, with our current method, identify the specific metabolites that differed across elevations or among sites, this is a promising area of future research. In addition, the absence of an elevational pattern in the diversity of these amphibian-associated bacterial communities seems to be somewhat consistent with observations of free-living soil bacterial communities, where no consistent elevational patterns have been observed. For example, one study showed a negative relationship between elevation and diversity within the phyla Acidobacteria in the western USA [66], another one found a peak in OTU richness at mid-elevation at Mount Fuji in Japan [67], and lastly, a study conducted in the Andean region showed no elevational trend in the bacterial communities from organic and mineral soils [68]. Additionally, although an understanding of the variation in the function of animal-associated bacterial communities across elevations is scarce, in free-living soil microbial communities, changes in environmental factors across elevations were associated with changes in community function, therefore influencing microbe-mediated nitrogen dynamics [69].
While the structure of bacterial communities on S. flotator was also relatively consistent among sites, with the exception of LowCope, there was variation across sites in metabolite profiles. In addition, the bacterial communities and metabolite profiles were not correlated either within elevations or across sites. Taken together, these results suggest that similar communities may produce different combinations of secondary metabolites at different sites, which could infer a potential functional plasticity in these bacterial communities. This could be mediated by differences in environmental conditions among sites. For instance, Daskin et al. [70] determined that specific bacterial isolates cultivated at a range of temperatures ecologically relevant to the amphibian-Bd interaction produce different quantities of the same metabolite and/or different types of metabolites. Moreover, Woodhams et al. [71] found variation in antifungal activity of metabolites against Bd from a bacterial isolate cultivated across a range of temperatures. Temperature is one of the main factors influencing bacterial cell growth and metabolic activity [72] and can influence the production of antibiotic compounds in bacteria [73, 74], potentially because of variation in temperature thresholds for the expression of genes involved in metabolite production [70, 75].
Understanding the link between biodiversity and ecosystem function can be challenging. Distinct approaches—studies across gradients and diversity manipulation—often give inconsistent results, including bacterial systems [76]. Nevertheless, it seems likely that the link between bacterial community structure and function across ecological systems may be influenced by environmental conditions, including both abiotic and biotic factors. For example, the diversity of free living soil ammonia- and nitrite-oxidizing bacteria changes across gradients of inorganic nitrogen availability, which influences the nitrification process [76–78]. Also, pathogen protection of corals by coral-associated bacteria is influenced by temperature [28]. In our system, it is possible that variation in environmental factors at different elevations, such as temperature, has more of an effect on bacterial community function (metabolite profiles) than on the structure of the communitites. Biotic factors that can impact host-associated microbial communities, in particular, include potential host pathogens. We could not examine the link between current pathogen infection with Bd and skin bacterial community structure or function in the present study due to the low incidence of Bd in our samples. However, prior experimental studies in other amphibian host species suggest that the bacterial community on the skin can potentially alter the disease outcome of Bd exposure and that Bd exposure can drive changes in the bacterial community structure [14, 57, 79–81]. Thus, potential context dependency driven by biotic factors, such as pathogen presence, might add another layer of complexity in understanding environmental context-dependent outcomes between microbes and their host and the mechanisms driving community function in these systems.
While we could not examine the effect of current Bd infection on the microbiome in our study, we did observe a longitudinal trend in alpha diversity of the skin bacterial communities across our sites that is consistent with the west to east spread of Bd in Panamá [36, 37, 82, 83]. Richness and phylogenetic diversity, in particular, decreased from the western sites to the eastern sites. Two prior studies have examined changes in the skin bacterial communities across host populations with different Bd histories in Panamá. Rebollar et al. [84] found that the terrestrial species Craugastor fitzingeri had lower skin bacterial community diversity in populations where Bd occurs at an endemic infection stage compared to Bd-naïve populations and higher relative abundance of some OTUs from the genera Cellulomonas, Sanguibacter, and Pseudomonas in the populations where Bd is endemic. However, in a separate study, across a gradient of Bd endemic sites, no clear west-east pattern was identified in the skin bacterial communities and metabolite profiles of two pond-breeding frogs, Agalychnis callidryas and Dendropsophus ebraccatus [14]. It may be that there are host species-specific patterns in the response of the skin microbiota to the presence of Bd in the environment. Species-specific responses of skin bacterial communities to Bd might add complexity to the studies addressing the ecological processes shaping the composition, structure, and function of these communities. This could partially explain the variable results regarding skin bacterial communities across Bd exposure experiments and across field studies working on distinct species [14, 57, 79–81].
We found that a very small fraction (0.7%) of the OTUs found on S. flotator can be considered as members of the core microbiome. But overall, the community was similar to what has been seen previously in Panamanian amphibians [14]. The most abundant OTUs identified in the S. flotator core belong to the genera Cellulomonas and Pseduomonas, which have also been found in high relative abundance on C. fitzingeri occurring at sites in Panamá where Bd is endemic [84]. Moreover, the families Comamonadaceae and Pseudomonadaceae, both abundant in the core microbiome of S. flotator, include many anti-Bd isolates from non-Bd-susceptible Panamanian amphibian species [15], and one OTU from the family Comamonadaceae was associated with individuals of the endangered species Atelopus zeteki that cleared experimental Bd infections [57]. Two core OTUs from S. flotator were in the family Enterobacteriaceae and were significantly more abundant in the Cope lowlands (LowCope), the only site with distinct bacterial communities relative to other sites. Interestingly, unlike the other study sites, the LowCope site was located in the proximity of a village. Importantly, there is evidence that amphibians and reptiles living on aquatic environments exposed to human domestic and industrial waste can harbor Enterobacteriaceae with high resistance against antibiotics and heavy metals, including the genus Citrobacter [85], which was identified in the present study.
Overall, we provide evidence that host-associated bacterial communities can be dynamic and that it is likely that their performance on the host depends on environmental conditions. In addition, we identified possible functional plasticity of amphibian skin bacteria along a gradient in the tropics where Bd epidemics have varied in intensity, though it would be premature to conclude that the observed variation in metabolite profiles, which we used to assess potential function, represents variation in function against Bd specifically. Considering the minimal variation in community structure of these bacterial communities across sites, we suggest that further characterization of the functions of these communities could be a promising area of research to elucidate how diversity influences function across environmental gradients. Lastly, we suggest that probiotic-based amphibian conservation efforts should consider the context-dependent function of probiotic candidates within the range of environmental conditions associated with sites of future reintroductions.
References
McFall-Ngai M, Hadfield MG, Bosch TCG, et al. (2013) Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U. S. A. 110:3229–3236. doi:10.2307/42583580?ref=search-gateway:7b87d5838b0343fae5e2fa5e5b3a74e2
Kaltenpoth M, Strupat K, Svatos A (2015) Linking metabolite production to taxonomic identity in environmental samples by (MA)LDI-FISH. ISME J 10:527–531. doi:10.1038/ismej.2015.122
Rebollar EA, Antwis RE, Becker MH, et al. (2016) Using “omics” and integrated multi-omics approaches to guide probiotic selection to mitigate chytridiomycosis and other emerging infectious diseases. Front. Microbiol. 7:733–719. doi:10.3389/fmicb.2016.00068
Wellington EM, Berry A, Krsek M (2003) Resolving functional diversity in relation to microbial community structure in soil: exploiting genomics and stable isotope probing. Curr. Opin. Microbiol. 6:295–301. doi:10.1016/S1369-5274(03)00066-3
Muegge BD, Kuczynski J, Knights D, et al. (2011) Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332:970–974. doi:10.1126/science.1198719
Harris RN, Lauer A, Simon MA, et al. (2009) Addition of antifungal skin bacteria to salamanders ameliorates the effects of chytridiomycosis. Dis. Aquat. Org. 83:11–16. doi:10.3354/dao02004
Hoyt JR, Cheng TL, Langwig KE, et al. (2015) Bacteria isolated from bats inhibit the growth of Pseudogymnoascus destructans, the causative agent of white-nose syndrome. PLoS One 10:e0121329–e0121312. doi:10.1371/journal.pone.0121329
Fraune S, Anton-Erxleben F, Augustin RE, et al. (2014) Bacteria-bacteria interactions within the microbiota of the ancestral metazoan Hydra contribute to fungal resistance. ISME J 9:1543–1556. doi:10.1038/ismej.2014.239
Rosenberg E, Koren O, Reshef L, et al. (2007) The role of microorganisms in coral health, disease and evolution. Nat Rev Micro 5:355–362. doi:10.1038/nrmicro1635
Sarmiento-Ramírez JM, van der Voort M, Raaijmakers JM, Diéguez-Uribeondo J (2014) Unravelling the microbiome of eggs of the endangered sea turtle Eretmochelys imbricata identifies bacteria with activity against the emerging pathogen fusarium falciforme. PLoS One 9:e95206–e95208. doi:10.1371/journal.pone.0095206
Flórez LV, Biedermann PHW, Engl T, Kaltenpoth M (2015) Defensive symbioses of animals with prokaryotic and eukaryotic microorganisms. Nat. Prod. Rep. 32:904–936. doi:10.1039/C5NP00010F
Walke JB, Becker MH, Loftus SC, et al. (2014) Amphibian skin may select for rare environmental microbes. ISME J 8:2207–2217. doi:10.1038/ismej.2014.77
Kueneman JG, Parfrey LW, Woodhams DC, et al. (2013) The amphibian skin-associated microbiome across species, space and life history stages. Mol. Ecol. 23:1238–1250. doi:10.1111/mec.12510
Belden LK, Hughey MC, Rebollar EA, et al. (2015) Panamanian frog species host unique skin bacterial communities. Front. Microbiol. 6:32–21. doi:10.3389/fmicb.2015.01171
Becker MH, Walke JB, Murrill L, et al. (2015) Phylogenetic distribution of symbiotic bacteria from Panamanian amphibians that inhibit growth of the lethal fungal pathogen Batrachochytrium dendrobatidis. Mol. Ecol. 24:1628–1641. doi:10.1111/mec.13135
Harris RN, James TY, Lauer A, et al. (2006) Amphibian pathogen Batrachochytrium dendrobatidis is inhibited by the cutaneous bacteria of amphibian species. EcoHealth 3:53–56. doi:10.1007/s10393-005-0009-1
Harris RN, Brucker RM, Walke JB, et al. (2009) Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J 3:818–824. doi:10.1038/ismej.2009.27
Becker MH, Brucker RM, Schwantes CR, et al. (2009) The bacterially produced metabolite Violacein is associated with survival of amphibians infected with a lethal fungus. Appl. Environ. Microbiol. 75:6635–6638. doi:10.1128/AEM.01294-09
Flechas SV, Sarmiento C, Cárdenas ME, et al. (2012) Surviving chytridiomycosis: differential anti-Batrachochytrium dendrobatidis activity in bacterial isolates from three lowland species of Atelopus. PLoS One 7:e44832–e44837. doi:10.1371/journal.pone.0044832
Brucker RM, Harris RN, Schwantes CR, et al. (2008) Amphibian chemical defense: antifungal metabolites of the microsymbiont Janthinobacterium lividum on the salamander Plethodon cinereus. J. Chem. Ecol. 34:1422–1429. doi:10.1007/s10886-008-9555-7
Brucker RM, Baylor CM, Walters RL, et al. (2007) The identification of 2,4-diacetylphloroglucinol as an antifungal metabolite produced by cutaneous bacteria of the salamander Plethodon cinereus. J. Chem. Ecol. 34:39–43. doi:10.1007/s10886-007-9352-8
Daszak P, Berger L, Cunningham AA, et al. (1999) Emerging infectious diseases and amphibian population declines. Emerging Infect Dis 5:735–748. doi:10.3201/eid0506.990601
Olson DH, Aanensen DM, Ronnenberg KL, et al. (2013) Mapping the global emergence of Batrachochytrium dendrobatidis, the amphibian chytrid fungus. PLoS One 8:e56802–e56813. doi:10.1371/journal.pone.0056802
Bronstein JL, Wilson WG, Morris WF (2003) Ecological dynamics of mutualist/antagonist communities. Am. Nat. 162:S24–S39. doi:10.1086/378645
Agrawal AA, Ackerly DD, Adler F, et al. (2007) Filling key gaps in population and community ecology. Front. Ecol. Environ. 5:145–152. doi:10.1890/1540-9295(2007)5[145:FKGIPA]2.0.CO;2
Hopkins SR, Wojdak JM, Belden LK (2016) Defensive symbionts mediate host–parasite interactions at multiple scales. Trends Parasitol.:1–12. doi:10.1016/j.pt.2016.10.003
Hofstetter R, Dempsey T, Klepzig K, Ayres M (2007) Temperature-dependent effects on mutualistic, antagonistic, and commensalistic interactions among insects, fungi and mites. Community Ecology 8:47–56. doi:10.1556/ComEc.8.2007.1.7
Shnit-Orland M, Kushmaro A (2009) Coral mucus-associated bacteria: a possible first line of defense. FEMS Microbiol. Ecol. 67:371–380. doi:10.1111/j.1574-6941.2008.00644.x
Kushmaro A, Loya Y, Fine M, Rosenberg E (1996) Bacterial infection and coral bleaching. doi: 10.1038/380396a0
Boyles JG, Willis CK (2010) Could localized warm areas inside cold caves reduce mortality of hibernating bats affected by white-nose syndrome? Front. Ecol. Environ. 8:92–98. doi:10.1890/080187
James TY, Toledo LF, Rödder D, et al. (2015) Disentangling host, pathogen, and environmental determinants of a recently emerged wildlife disease: lessons from the first 15 years of amphibian chytridiomycosis research. Ecol Evol 5:4079–4097. doi:10.1002/ece3.1672
Daskin JH, Alford RA (2012) Context-dependent symbioses and their potential roles in wildlife diseases. Proceedings of the Royal Society B: Biological Sciences 279:1457–1465. doi: 10.1098/rspb.2011.2276
Crawford AJ, Lips KR, Bermingham E, Wake DB (2010) Epidemic disease decimates amphibian abundance, species diversity, and evolutionary history in the highlands of central Panama. Proc. Natl. Acad. Sci. U. S. A. 107:13777–13782. doi:10.2307/25708801?ref=search-gateway:c5e8aac1656df05dfd381d5c42b5c4df
Lips KR, Brem F, Brenes R, et al. (2006) Emerging infectious disease and the loss of biodiversity in a Neotropical amphibian community. Proceedings of the National Academy of Sciences 103:3165–3170. doi: 10.1073/pnas.0506889103
Ryan MJ, Lips KR, Eichholz MW (2008) Decline and extirpation of an endangered Panamanian stream frog population (Craugastor punctariolus) due to an outbreak of chytridiomycosis. Biol. Conserv. 141:1636–1647. doi:10.1016/j.biocon.2008.04.014
Woodhams DC, Kilburn VL, Reinert LK, et al. (2008) Chytridiomycosis and amphibian population declines continue to spread eastward in Panama. EcoHealth 5:268–274. doi:10.1007/s10393-008-0190-0
Rebollar EA, Hughey MC, Harris RN, et al. (2014) The lethal fungus Batrachochytrium dendrobatidis is present in lowland tropical forests of far eastern Panamá. PLoS One 9:e95484–e95488. doi:10.1371/journal.pone.0095484
Sapsford SJ, Alford RA, Schwarzkopf L (2013) Elevation, temperature, and aquatic connectivity all influence the infection dynamics of the amphibian chytrid fungus in adult frogs. PLoS One. doi:10.1371/journal.pone.0082425
Catenazzi A, Lehr E, Vredenburg VT (2013) Thermal physiology, disease, and amphibian declines on the eastern slopes of the Andes. Conserv. Biol. 28:509–517. doi:10.1111/cobi.12194
Gründler MC, Toledo LF, Parra-Olea G, et al. (2012) Interaction between breeding habitat and elevation affects prevalence but not infection intensity of Batrachochytrium dendrobatidis in Brazilian anuran assemblages. Dis. Aquat. Org. 97:173–184. doi:10.3354/dao02413
Kriger KM, Hero J-M (2008) Altitudinal distribution of chytrid (Batrachochytrium dendrobatidis) infection in subtropical Australian frogs. Austral Ecology 33:1022–1032. doi:10.1111/j.1442-9993.2008.01872.x
Brem F, Lips KR (2008) Batrachochytrium dendrobatidis infection patterns among Panamanian amphibian species, habitats and elevations during epizootic and enzootic stages. Dis. Aquat. Org. 81:189–202. doi:10.3354/dao01960
Jaramillo AC, Wilson LD, Ibáñez R, Jaramillo F (2010) The herpetofauna of Panama: distribution and conservation status. Conservation of Mesoamerican amphibians and reptiles. Eagle Mountain Press, Eagle Mountain, pp. 604–671
Lauer A, Simon MA, Banning JL, et al. (2007) Common cutaneous bacteria from the eastern red-backed salamander can inhibit pathogenic fungi. Copeia 630–640
Umile TP, McLaughlin PJ, Johnson KR, et al. (2014) Nonlethal amphibian skin swabbing of cutaneous natural products for HPLC fingerprinting. Anal. Methods 6:3277–3284. doi:10.1039/C4AY00566J
Lauber CL, Zhou N, Gordon JI, et al. (2010) Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol. Lett. 307:80–86. doi:10.1111/j.1574-6968.2010.01965.x
Whitfield SM, Kerby J, Gentry LR, Donnelly MA (2012) Temporal variation in infection prevalence by the amphibian chytrid fungus in three species of frogs at La Selva, Costa Rica. Biotropica 44:779–784. doi:10.1111/j.1744-7429.2012.00872.x
Caporaso JG, Lauber CL, Walters WA, et al. (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi:10.1038/ismej.2012.8
Aronesty E (2011) ea-utils: Command-line tools for processing biological sequencing data. Expression Analysis 1–1
Caporaso JG, Kuczynski J, Stombaugh J, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7:335–336. doi:10.1038/nmeth.f.303
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi:10.1093/bioinformatics/btq461
DeSantis TZ, Hugenholtz P, Larsen N, et al. (2006) Greengenes, a chimera-checked 16S rRNA Gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072. doi:10.1128/AEM.03006-05
Caporaso JG, Bittinger K, Bushman FD, et al. (2010) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi:10.1093/bioinformatics/btp636
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267. doi:10.1128/AEM.00062-07
Bokulich NA, Subramanian S, Faith JJ, et al. (2012) Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Meth 10:57–59. doi:10.1038/nmeth.2276
Boyle DG, Boyle DB, Olsen V, et al. (2004) Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis. Aquat. Org. 60:141–148. doi:10.3354/dao060141
Becker MH, Walke JB, Cikanek S, et al. (2015) Composition of symbiotic bacteria predicts survival in Panamanian golden frogs infected with a lethal fungus. Proceedings of the Royal Society B: Biological Sciences 282:20142881–20142881. doi: 10.1098/rspb.2014.2881
R Core Team (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Bates D, Maechler M, Bolker BM, Walker SC (2014) Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67:1–48
Venables WN, Ripley BD (2002) Modern applied statistics with S. Fourth Edition. 1–1
Hothorn T, Bretz F, Westfall P (2008) Simultaneous inference in general parametric models. Biom. J. 50:346–363. doi:10.1002/bimj.200810425
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecology 26:32–46. doi:10.1111/j.1442-9993.2001.01070.pp.x
Okansen J, Blanchet FG, Kindt R, et al. (2016) vegan: community ecology package. R package version 2.3–5 https://CRAN.R-project.org/package=vegan. 1–1
Shade A, Handelsman J (2011) Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14:4–12. doi:10.1111/j.1462-2920.2011.02585.x
Warnes GR, Bolker B, Bonebakker L, et al. (2016) gplots: various R programming tools for plotting data. R package version 3.0.1 https://CRAN.R-project.org/package=gplots. 1–1
Bryant JA, Lamanna C, Morlon H, et al. (2008) Colloquium paper: microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proc. Natl. Acad. Sci. U. S. A. 105(Suppl 1):11505–11511. doi:10.1073/pnas.0801920105
Singh D, Takahashi K, Kim M, et al. (2012) A hump-backed trend in bacterial diversity with elevation on Mount Fuji, Japan. Microb. Ecol. 63:429–437. doi:10.1007/s00248-011-9900-1
Fierer N, McCain CM, Meir P, et al. (2011) Microbes do not follow the elevational diversity patterns of plants and animals. Ecology 92:797–804
Yang Y, Gao Y, Wang S, et al. (2014) The microbial gene diversity along an elevation gradient of the Tibetan grassland. ISME J 8:430–440. doi:10.1038/ismej.2013.146
Daskin JH, Bell SC, Schwarzkopf L, Alford RA (2014) Cool temperatures reduce antifungal activity of symbiotic bacteria of threatened amphibians—implications for disease management and patterns of decline. PLoS One 9:e100378–e100379. doi:10.1371/journal.pone.0100378
Woodhams DC, Brandt H, Baumgartner S, et al. (2014) Interacting symbionts and immunity in the amphibian skin mucosome predict disease risk and probiotic effectiveness. PLoS One 9:e96375–e96313. doi:10.1371/journal.pone.0096375
Gacheva G, Gigova L (2014) Biological activity of microalgae can be enhanced by manipulating the cultivation temperature and irradiance. Open Life Sciences 9:1–14. doi:10.2478/s11535-014-0350-x
Humair B, González N, Mossialos D, et al. (2009) Temperature-responsive sensing regulates biocontrol factor expression in Pseudomonas fluorescens CHA0. ISME J 3:955–965. doi:10.1038/ismej.2009.42
Raaijmakers JM, Vlami M, de Souza JT (2002) Antibiotic production by bacterial biocontrol agents. Antonie Van Leeuwenhoek 81:537–547
Cundliffe E (2006) Antibiotic production by actinomycetes: the Janus faces of regulation. J. Ind. Microbiol. Biotechnol. 33:500–506. doi:10.1007/s10295-006-0083-6
Krause S, Le Roux X, Niklaus PA (2014) Trait-based approaches for understanding microbial biodiversity and ecosystem functioning. Salamandra 5:1–10. doi:10.3389/fmicb.2014.00251/abstract
Webster G, Embley TM, Freitag TE (2005) Links between ammonia oxidizer species composition, functional diversity and nitrification kinetics in grassland soils. Salamandra 7:676–684. doi:10.1111/j.1462-2920.2004.00740.x
Attard E, Poly F, Commeaux C, et al. (2010) Shifts between Nitrospira- and Nitrobacter-like nitrite oxidizers underlie the response of soil potential nitrite oxidation to changes in tillage practices. Environ. Microbiol. 12:315–326. doi:10.1111/j.1462-2920.2009.02070.x
Walke JB, Becker MH, Loftus SC, et al. (2015) Community structure and function of amphibian skin microbes: an experiment with bullfrogs exposed to a chytrid fungus. PLoS One 10:e0139848. doi:10.1371/journal.pone.0139848
Jani AJ, Briggs CJ (2014) The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proceedings of the National Academy of Sciences 111:E5049–E5058. doi: 10.1073/pnas.1412752111
Bresciano JC, Salvador CA, Paz-y-Miño C, et al. (2015) Variation in the presence of anti-Batrachochytrium dendrobatidis bacteria of amphibians across life stages and elevations in Ecuador. EcoHealth 12:310–319. doi:10.1007/s10393-015-1010-y
Lips KR, Diffendorfer J, Mendelson JR, Sears MW (2008) Riding the wave: reconciling the roles of disease and climate change in amphibian declines. PLoS Biol. 6:e72–e14. doi:10.1371/journal.pbio.0060072
Rodríguez-Brenes S, Rodríguez D, Ibáñez R, Ryan MJ (2016) Spread of amphibian chytrid fungus across lowland populations of Túngara frogs in Panamá. PLoS One 11:e0155745–e0155748. doi:10.1371/journal.pone.0155745
Rebollar EA, Hughey MC, Medina D, et al. (2016) Skin bacterial diversity of Panamanian frogs is associated with host susceptibility and presence of Batrachochytrium dendrobatidis. ISME J 10:1682–1695. doi:10.1038/ismej.2015.234
Hacioglu N, Tosunoglu M (2013) Determination of antimicrobial and heavy metal resistance profiles of some bacteria isolated from aquatic amphibian and reptile species. Environ. Monit. Assess. 186:407–413. doi:10.1007/s10661-013-3385-y
Acknowledgements
Thanks to Molly Bletz for assistance in the field, Skylar Hopkins and James Skelton for statistical advice, Eria Rebollar for help with the export and import of samples, and Jennifer Smith for producing Fig. 1. We also thank the Smithsonian Tropical Research Institute for providing logistical support and laboratory space and the Interfaces of Global Change graduate program at Virginia Tech for fellowship support to DM. Funds were provided by the National Science Foundation (NSF: DEB-1136640 to LKB; DEB-1136662 to KPCM). Samples were collected under the scientific permit SE/A-35-13 granted by MIAmbiente.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Medina, D., Hughey, M.C., Becker, M.H. et al. Variation in Metabolite Profiles of Amphibian Skin Bacterial Communities Across Elevations in the Neotropics. Microb Ecol 74, 227–238 (2017). https://doi.org/10.1007/s00248-017-0933-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-017-0933-y