
Abstract
Unraveling the distribution patterns of plants and animals along the elevational gradients has been attracting growing scientific interests of ecologists, whether the microbial communities exhibit similar elevational patterns, however, remains largely less documented. Here, we investigate the biogeographic distribution of soil archaeal and bacterial communities across three vertical climate zones (3,106–4,479 m.a.s.l.) in Mt. Shegyla on the Tibetan Plateau, by combining quantitative PCR and high-throughput barcoded pyrosequencing approaches. Our results found that the ratio of bacterial to archaeal 16S rRNA gene abundance was negatively related with elevation. Acidobacteria dominated in the bacterial communities, Marine benthic group A dominated in the archaeal communities, and the relative abundance of both taxa changed significantly with elevation. At the taxonomic levels of domain, phylum, and class, more bacterial taxa than archaeal exhibited declining trend in diversity along the increasing elevational gradient, as revealed by Shannon and Faith’s phylogenetic diversity indices. Unweighted UniFrac distance clustering showed that the bacterial communities from the mountainous temperate zone clustered together, whereas those from the subalpine cool temperate zone clustered together. However, the partitioning effect of elevational zones on the archaeal community was much weaker compared to that on bacteria. Redundancy analysis revealed that soil geochemical factors explained 58.3 % of the bacterial community variance and 75.4 % of the archaeal community variance. Taken together, we provide evidence that soil bacteria exhibited more apparent elevational zonation feature and decreased diversity pattern than archaea with increasing elevation, and distribution patterns of soil microbes are strongly regulated by soil properties along elevational gradient in this plateau montane ecosystem.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since Alexander von Humboldt’s journey off the Chimborazo volcano in Ecuador in 1802, global ecologists have been exploring the montane ecosystem for over 200 years. Two important features of montane ecosystem have been well documented through these studies. One is the altitudinal belts of community in montane ecosystems. For instance, environmental factors including temperature, precipitation, solar radiation, etc. experience dramatic changes over short spatial distances from the baseband to the top of the mountain [1]. These changes could collectively lead to formation of natural climate gradients regulating biodiversity succession and organism distributions [2]. In this case, altitudinal zonation of macro communities has been widely observed along the elevational gradient [3]. For example, cryptogamic plant communities along an altitudinal gradient showed obvious zonation in tropical Andean rain forests [4], and altitudinal zonation of bird communities was observed along a 1,550-m forest gradient in the Udzungwa Mountains in Tanzania [5]. However, as for the microorganisms, despite the strong influences of environmental factors on their community structures [6] and the significant importance of microbes in ecosystem functioning and primary production, it is largely unknown how these microbes are structured by elevations, and limited studies have been conducted to provide microbial community composition patterns in montane ecosystems.
Another important feature of montane ecosystem is the changing biological diversity with elevation. There has been a large body of studies addressing how the biodiversity of plant and animal changes along the elevations in montane ecosystems. Most of these investigations, however, focused on the macroorganisms and had reported a variety of contrasting biodiversity elevation patterns, for instance, a pattern of decline [7], a unimodal pattern [8], and a pattern of increase [9]. However, the diversity pattern of microorganisms along the elevational gradients was not observed until an investigation using modern molecular approaches was carried out on the Rocky Mountains across a 920-m elevational gradient from 2,460 to 3,880 m [10]. In this survey, Bryant et al. found that soil Acidobacteria exhibited a declining pattern of phylogenetic diversity with increasing altitude, whereas Angiosperm showed a typical unimodal pattern. Another comparative study between the diversity patterns of macroorganism and microorganism along an elevational gradient from 200 to 3,450 m in the eastern Peru montane areas observed no clear pattern for the bacterial diversity, whereas plants and animals exhibited a significant pattern of decline [11]. In addition, a significant unimodal pattern of soil bacterial diversity along an elevational gradient was reported on the Mt. Fuji from 1,000 to 3,700 m recently [12].
Quite a few hypothesis and theories have been proposed to uncover the mechanisms that drive those elevational biological diversity patterns. For example, a mid-domain effect (MDE) was widely used in the interpretation of unimodal diversity pattern in macro ecology, which emphasizes the geometric constriction upon species distribution, and proposes that species usually accumulate in the middle areas along elevation [13]. However, applicability of MDE in explaining microbial distribution was doubtable when the measurement of dispersal limits for microbes is still ambiguous [14]. In a research on freshwater habitat, the elevational diversity pattern of microbes was better explained by environmental factors rather than by MDE [15]. The decreased pattern of soil Acidobacteria on Rocky Mountains, as reported by Bryant et al. [10], also found to be closely related to environmental factors. These findings highlight the role of environmental factors in structuring elevational microbial distribution patterns, suggesting that related theory like Bass-Becking hypothesis might be indicative in interpreting microbial distribution patterns in montane ecosystems.
Despite of the available knowledge, we are still unclear about the altitudinal distribution patterns of microbial communities when considering the differences in sampling and analytical methodologies across different investigations. Comparative studies are common in biogeography; however, most of microbial biogeographic studies have focused merely on bacteria [11] or archaea [16]. In fact, both bacteria and archaea have been found to be remarkably abundant and functionally important in terrestrial ecosystems, and could contribute differently to ecosystem functioning and biogeochemical cycling [17]. Archaea and bacteria highly differ in their metabolic characteristics and genetic features [18], indicating that these two prokaryotic taxa may respond differently to environmental factors.
As the highest plateau in the world, the Tibetan Plateau is a unique habitat that characterizes by harsh climatic conditions including dry air, high UV, and low oxygen. These special conditions make the Tibetan Plateau a separate geographic region, resulting in the formation of particular flora and fauna [19, 20]. However, scant information is available concerning the biogeographic patterns and ecological characteristics of microbial communities in this typical plateau. On the other hand, the Tibetan Plateau is a vulnerable landscape that is highly sensitive to global climate change, which could greatly impact plant nutrition and ecosystem functioning by inevitably affecting the distribution of microorganisms [21]. Soil microbes have been recognized as a keystone in the global cycling of nitrogen and carbon, and a better understanding of their distribution characteristics along the elevational gradients would strengthen our ability to manage montane ecosystems and to mitigate the impacts of global climate changes (e.g., elevated atmospheric temperature and greenhouse gases) [22] by developing targeted options on soil microorganisms.
In this study, we examined the altitudinal distribution of soil bacteria and archaea across three climate zones along a montane forest landscape on the Tibetan Plateau. We aimed to determine whether soil bacteria and/or archaea exhibit clear elevational patterns in this plateau montane ecosystem. We hypothesized that (1) similar to macroorganisms, soil archaea and bacteria might also show certain distribution patterns along the altitude elevations because of the remarkable altitudinal shifts in environmental factors in montane ecosystems; (2) archaeal and bacterial taxa might have divergent distribution patterns due to their highly different genetic features and inconsistent metabolic characteristics affected by environmental factors.
Materials and Methods
Site Description and Soil Sampling
Mount Shegyla, an offshoot of Mt. Nyainqêntanglha, is characterized by a plateau landscape that lies in Nyingchi (94°25′-94°45′ E, 29°35′-29°57′ N) in Southeast Tibet, China. We investigated here because there are very typical high plateau landscapes on this mountain. Along the elevational gradient, there are apparent mountainous temperate zones between 2,700 and 3,400 m, subalpine cool temperate zones between 3,400 and 4,300 m, and alpine frigid zones above 4,300 m. Aboveground plant species in these zones are highly different: Quercus semecarpifolia and Abies are abundant in mountainous temperate zones, Aibes georgei var. smithii is abundant in subalpine cool temperate zones, and Rhododendron and Sabina saltuaria are abundant in alpine frigid zones. Soil samples were collected across the three vertical climate zones in July 2011. Six elevational sites with representative aboveground vegetation were identified ranging from 3,118 to 4,477 m across the elevational zones. We did not sample from the baseband because of the obvious anthropogenic disturbance there. At each site, we collected three soil samples from three separated 10 m × 10 m plots, by pooling eight upper 10-cm soil cores randomly taken from every plot. Detailed information about these soil samples is listed in Table 1. For each sample, 500 g of soil sieved to 2 mm was stored at 4 °C for physicochemical analyses, and 50 g of soil was encapsulated in a plastic tube and kept at −80 °C before DNA extraction.
Soil Physicochemical Analyses and DNA Extraction
Soil texture was evaluated using a Mastersizer 2000 Laser Diffraction Particle Analyzer (Malvern Instruments Ltd, Malvern, UK) according to the universal criteria of soil particle size (clay <0.002 mm, silt 0.02–0.002 mm, sand >0.02 mm). Exchangeable cations were extracted using 1 M neutral ammonium acetate solution and quantified through the Kjeldahl method to measure the cation exchange capacity (CEC). Soil pH was determined at a ratio of 1:2.5 (soil to water, w/w). Soil organic carbon (SOC) was determined by the K2Cr2O7 oxidation method mentioned by Cao et al. [23]. Total nitrogen (TN) was determined using a Vario EL III analyzer (Elementar Analysensysteme GmbH, Hanau, Germany).
The total genomic DNA was isolated from 0.25 g of soil using the MOBIO Ultraclean Soil DNA Isolation Kit (MOBIO laboratories, Carlsbad, California, USA) as per the manufacturer’s instructions, with slight modifications that the filters were washed twice with the solution C5. The extracted DNA was stored at −80 °C, and tenfold diluted DNA was used in the downstream PCR and quantitative PCR analyses.
16S rRNA Gene Amplification and Barcoded Pyrosequencing
Quantitative PCR assays were carried out on a BIO-RAD IQ5 thermal cycler (Bio-Rad Laboratories, Inc., California, USA) to determine the prokaryotic abundance. A TaqMan probe TM1389F was employed to quantify the bacterial 16S rRNA gene by using the primer pairs 1369 F/1492R, the 3′ end of which was marked as TAMRA, whereas the 5′ end was marked by FAM [24]. The 25-μl PCR reactions contained 12.5 μl Premix Ex TaqTM (Takara Biotechnology, Dalian, China), 2.5 μM of each primer, 1.5 μM of the Taqman probe, and a 1-μl DNA template. The abundance of the archaeal 16S rRNA gene was measured using the primer pairs 364 F/934R [25] according to the SYBR® TAKARA Premix Ex Taq manual (Takara Biotechnology).
A barcoded pyrosequencing procedure targeting the 16S rRNA gene of bacteria and archaea was employed to decipher the prokaryotic community composition. The primer pair 27 F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 519R (5′-GWATTACCGCGGCKGCTG-3′) was used to amplify the V1–V3 region of the bacterial 16S rRNA gene [26], and the primer pair 364F (5′-CGGGGYGCASCAGGCGCGAA-3′) and 934R (5′-GTGCTCCCCCGCCAATTCCT-3′) to amplify the V3–V6 region of the archaeal 16S rRNA gene [25]. Two Roche-provided adaptors, a Primer A-Key (5′-CGTATCGCCTCCCTCGCGCCATCAG-3′) and a Primer B-Key (5′-CTATGCGCCTTGCCAGCCCGCTCAG-3′), were added to the 5′ end of the forward primer and reverse primer, respectively. A 10-bp MID sequence was inserted between the Primer A-Key and the forward primer for barcode identification. The 25-μl PCR reaction system consisted of 12.5 μl Premix Ex Taq™ Version 2.0 (Takara Biotechnology), 1 μl forward primer (10 μM) and 1 μl reverse primer (10 μM), 2 μl DNA template (1–10 ng), and 8.5 μl sterilized H2O. A Wizard SV Gel and PCR Clean-Up System (Progema Corporation, Madison, USA) was used in the purification of PCR products, according to the manufacturer’s instructions. The purified PCR products were equimolarly mixed in a 2-ml tube. The sequencing was carried out on a 454 GS FLX Titanium pyrosequencer (Roche 454 Life Sciences, Branford, CT, USA).
Processing of the Pyrosequencing Data
Raw sequences that obtained through high-throughput barcoded pyrosequencing were processed following the Quantitative Insights into Microbial Ecology (QIIME) pipeline [27]. Prior to clustering, reads with quality score lower than 20, improper primers, or ambiguous bases were discarded. The FASTA files of all samples were combined together through an add_qiime_labels.py script. We selected PyNAST as the alignment algorithm [28] against a 13_8 Greengenes database [29]. Chimeras were identified and eliminated via performing the identify_chimeric_seqs.py and filter_fasta.py scripts within QIIME. The resultant high-quality sequences that shared at least 97 % similarity were clustered into operational taxonomic units (OTUs) by using uclust [30]. Representative sequences from individual OTUs were classified by the RDP classifier [31] against the Greengenes taxonomic database with a confidence threshold of 0.8. The community compositions of bacteria and archaea were then described by the relative abundance of sequences that assigned to each taxon. At each taxonomic level, we included only the taxa that were observed in all of the samples. Taxa that occasionally observed in individual samples accounted for a minor fraction of total bacteria (<0.01 %) and were not included in the downstream assemblage analyses.
To eliminate the bias on diversity comparison caused by unevenly sequencing, 869 bacterial and 278 archaeal sequences per sample were stochastically picked out through a resampling step before the subsequent diversity calculation. The 97 % similarity was employed in the OTU definition for estimation of the alpha and beta diversity. The Shannon index and Faith’s phylogenetic index [32] were calculated to compare the alpha diversity. The unweighted UniFrac distance [33] was introduced as the beta diversity metrics. A taxa filtering script provided by QIIME was performed to separate the OTU tables of individual microbial taxa, according to which the diversity of specific taxa was analyzed. An Unweighted Pair Group Method with Arithmetic Mean clustering (UPGMA) procedure based on the unweighted UniFrac distance was performed in QIIME to compare the pair-wise community similarity.
Statistical Analysis
Spearman’s correlation analysis was performed to examine the relationships between altitude and soil physiochemical factors in SPSS 19 (IBM Co., Armonk, NY, USA). A permutation multivariate analysis of variance (PERMANOVA) was conducted to test the significance of community composition variance in R 3.0.1 [34] using the Vegan package [35]. Redundancy analysis was employed to measure the effects of soil geochemical factors on community structures, and a Monte Carlo test with 999 permutations was carried out to test the significance of relationship between community and environmental matrix. An envfit function in Vegan package was employed to exclude factors that have no significant effects on community structure from RDA plots. Relative contribution of each environmental variable was then distinguished using a distance-based linear modeling approach in R [36].
Results
Changes in Soil Geochemical Factors Along the Elevations on Mt. Shegyla
A very obvious trend of soil geochemical factors along the altitude elevations was observed. Among all the factors examined (Table 1), soil pH ranging from 4.68 to 6.43 behaved a significantly negative relationship with altitude (r = −0.541, p < 0.01). Soil organic carbon (r = 0.654, p < 0.01) and moisture content (r = 0.656, p < 0.01), on the contrary, showed a significantly positive relationship with altitude. The content of clay showed a significantly negative relationship with altitude (r = −0.488, p < 0.05). No significant relationships with altitude could be detected on total nitrogen (r = 0.476, p = 0.054), C/N ratio (r = 0.179, p = 0.492), or CEC (r = 0.280, p = 0.277).
Abundance and Community Compositions of Soil Bacteria and Archaea Assemblages on Mt. Shegyla
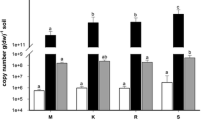
The log numbers of archaeal 16S rRNA gene copies (ranging from 7.82 to 8.43) was much lower than those of bacterial 16S rRNA gene copies (ranging from 10.04 to 10.93) in each gram of soil (Fig. 1). Archaeal abundance accounted for 1–13 % of the total prokaryotes (the sum of the bacterial and archaeal 16S rRNA gene copy numbers in individual samples). A significantly higher abundance of bacteria was observed at the elevational sites of 3,352 m compared to those in other elevations. Along the increasing elevation from 3,106 to 4,479 m, a significant negative relationship between altitude and the log ratio of bacterial to archaeal abundance was observed (r = −0.515, p < 0.05). However, no significant correlations between altitude and the bacterial (r = −0.341, p = 0.18) or archaeal abundance (r = 0.323, p = 0.21) could be observed.

Log numbers of 16S rRNA gene copies of soil bacteria and archaea and their ratio at different altitude gradient
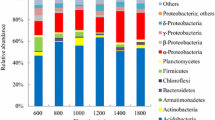
Across all the examined samples, the barcoded pyrosequencing yielded 96,965 high-quality 16S rRNA gene sequences in total. Of these sequences, 75 % could be classified into the bacteria domain, and the other 25 % could be affiliated within the archaea domain. On average, 4,279 bacterial sequences and 1,424 archaeal sequences were obtained for each sample. The bacterial and archaeal community compositions were profiled according to their relative abundance at the taxonomic levels of phylum and order (Fig. 2). Nearly 100 % of the amplicons generated by the primer set 27 F/519R hit the bacteria domain according to the Greengenes database. Among those taxa examined, Acidobacteria (37.8 %) and Proteobacteria (35.4 %) were the most dominant phyla of the bacterial community, and we noticed that the relative abundance of Acidobacteria was significantly higher in the elevational sites above 3,915 m. Planctomycetes (3.3 %), Actinobacteria (4.2 %), Bacteroidetes (2.9 %), Gemmatimonadetes (2.4 %), and Verrucomicrobia (1.8 %) were also found to be widely distributed in all of the samples. A total of 98.1 % of the archaeal sequences were classified into Crenarchaeota (73.7 %) and Euryarchaeota (24.3 %) as shown in Fig. 2b. At the taxonomic level of class, Marine benthic group A (MGBA, 62.1 %), Thaumarchaeota (10.4 %), and Thermoplasmata (18.8 %) dominate the archaeal community (Fig. 2b). The relative abundance of MBGA exhibited an apparent increasing trend with altitude before reaching the alpine frigid zones at 4,477 m.

Relative abundance of dominant phyla of bacteria (a) and archaea (b) along an elevational gradient. Unclassified_Cre indicates the unclassified Crenarchaeota
Diversity Patterns of the Bacterial and Archaeal Communities on Mt. Shegyla
To explore their elevational diversity patterns, we fitted the altitude curve with both Faith’s PD index and Shannon index of bacteria and archaea at different taxonomic levels (Table 2). Bacteria were determined at the levels of domain and phylum, and Proteobacteria were further analyzed at the class level because of its high relative abundance in total bacteria. Archaea were studied at the levels of domain, phylum, and class.
A pattern of decline in the bacterial community diversity along the increasing elevations could be detected at all taxonomic levels (Table 2). For instance, at the domain level, bacteria exhibited a declining pattern of phylogenetic diversity. At the level of phylum, Acidobacteria showed a pattern of decline in both diversity index. Planctomycetes showed a decreasing pattern via Faith PD, and Actinobacteria, Gemmatimonadetes, and Verrucomicrobia showed a declining pattern via Shannon index. At the class level, alpha-Proteobacteria and delta-Proteobacteria showed a declining diversity pattern, despite that no significant trend was observed in total Proteobacteria. Archaea did not show obvious diversity patterns at the domain level along the increasing elevations. At the levels of phylum and class, only Euryarchaeota and their dominant clade Thermoplasmata showed a declining pattern as revealed by Shannon index.
A UPGMA hierarchical clustering procedure was performed to compare the β-diversity of the bacterial and archaeal communities using the unweighted UniFrac distance. Three main clusters that were marked with different colors could be clearly identified for bacteria, which showed good consistence with the sample locations from different vertical climate zones (Fig. 3a). There were also three main clusters in the archaeal UPGMA dendrogram (Fig. 3b), but consistence between the locations and clusters of archaeal communities was much weaker than that of the bacterial communities. For both bacteria and archaea, we found that communities from the same elevational zones were more likely to be clustered together.

UPGMA dendrogram constructed from an unweighted UniFrac matrix of the bacterial (a) and archaeal (b) 16S rRNA gene sequences. Three main clusters colored with red, green, and blue are distinguished for both bacteria and archaea. The sources of communities are also listed as follows: T represents the mountainous temperate zones, C represents the subalpine cool temperate zones, and F represents the alpine frigid zones
A PERMAVONA test based on the unweighted UniFrac matrix was performed to further determine the significance of the divergence in bacterial and archaeal community compositions between different elevational zones. The bacterial communities could be clearly separated based on different sample sources (p < 0.01). Archaeal communities, although not as significant as bacteria, were also found to be significantly separated by the different elevational zones (p < 0.05).
Redundancy Analysis of Soil Physiochemical Factors and Community Structures
The influences of different soil geochemical factors on community assemblages were measured by redundancy analysis (Fig. 4). The first two axes accounted for 58.3 and 75.4 % of the variance for the bacterial (Fig. 4a) and archaeal (Fig. 4b) communities, respectively. Of all the soil geochemical factors examined, soil pH (43.7 %), clay contents (6.1 %), TN (3.2 %), and SOC (1.0 %) were found to be significant for the bacteria, while the archaeal community was found to be significantly affected by soil pH (52.9 %), clay contents (6.0 %), TN (1.9 %), SOC (3.2 %), CEC (0.2 %), and soil moisture (5.7 %) content. For the bacterial communities, soil pH was positively correlated with those from the temperate zone but negatively correlated with those from the cool temperate zone. A similar finding was observed for the archaeal communities, except that the soil moisture content and CEC positively affected those from the cool temperate zone.

Redundancy analysis of the environmental factors and community structures of bacteria (a) and archaea (b). The red spot represents communities from the mountainous temperate zones; the green square represents communities from the subalpine cool temperate zones; and the purple triangle represents communities from the alpine frigid zones. All of the displayed geochemical factors have passed a significance test. SOC indicates the soil organic carbon, CEC indicates the cation exchangeable capacity, and TN indicates the total nitrogen. The Ac indicates Actinobacteria; Ba indicates Bacteroidetes; Ge indicates Gemmatimonadetes; Pl indicates Planctomycetes; and Ve indicates Verrucomicrobia
Discussion
Altitudinal Distribution Patterns of Soil Microorganisms on Mt. Shegyla
In this investigation, by employing quantitative PCR and next-generation barcoded pyrosequencing, our study provided important evidence that soil bacterial and archaeal community compositions exhibited an altitudinal zonation feature on Mt. Shegyla, accompanied by apparent decreasing diversity patterns along the increasing elevation. These findings corresponded to our first hypothesis, indicating that elevational distribution patterns of soil microbes could also be observed in this high plateau montane ecosystem.
Since no unimodal pattern was detected for any bacterial or archaeal taxa, the MDE would not be suitable for interpreting the diversity patterns of soil microbes on Mt. Shegyla. A decreased diversity pattern, however, was observed on several microbial taxa at different taxonomic levels examined. Several researches reporting a decreased elevational pattern of microbes have emphasized the great importance of environmental variables [10]. We examined the correlations between the matrix of environmental variables and community structure, and found that the distribution patterns of soil microbes were closely related with environmental factors on Mt. Shegyla, indicating that the Bass-Becking hypothesis should be taken into consideration. Bass-Becking hypothesis, known as “everything is everywhere, but the environment selects,” assumed that microbes are ubiquitous, and environmental factors make decisive contributions to structuring microbial community compositions [37]. In our study, correlation analyses revealed that the soil geochemical factors exhibited an obvious distribution trend on Mt. Shegyla, and the redundancy analysis showed that they greatly influenced the microbial community compositions. Apparent diversity patterns of different microbial taxa highlighted the importance of these soil environmental factors in regulating microbial distribution.
The critical soil geochemical factors, as we showed in this study, highly varied across the elevational climate zones. Of all the factors we examined, soil pH, SOC, and TN gained more attention for their considerable influences on microbial community compositions and diversity patterns (Table S1). As the most important feature of soil, pH affected soil microbes not only by conditioning enzymes activity but also by constraining the availability of nutrients through determining the ionization equilibrium in soil. For instance, pH determined the community composition and biogeography of ammonia oxidizers by affecting ionization equilibrium of nitrate and ammonia in soils [17]. And pH-driven diversity patterns were observed horizontally [6] as well as along the elevational gradient [38]. As the fundamental energy sources and component elements for microorganisms, soil organic material and nitrogen also significantly affected the elevational distribution patterns of soil microbes via determining the metabolism of soil microbes [39]. In addition to the edaphic factors, climatic scenarios on the Tibetan Plateau should also be an important consideration. Effects of climate can be interpreted at both regional and local scales. At regional scale, harsh climatic conditions make the Tibetan Plateau a particular biogeographic region. For example, seasonal freezing and thawing is a very important ecological process in the Tibetan Plateau. Alternative freezing and thawing impact on soil microbial communities not only by directly affecting on the metabolic activity and reproduction of soil microbes but also by indirectly changing soil physical properties like temperate, moisture, and rock weathering that closely related with soil mechanical composition like clay content. At local scale, different climate zones can preserve over very short geographic distances because of the steep environmental gradient. The elevational climate zones structured the community of plant, and variation in plants could be an important factor that resulting into variable soil microbial communities especially in the rhizosphere [40].
In a biogeographic frame of environmental factors and historical contingencies, the distribution patterns of microbes were more susceptible to environmental factors at local scale (ecological process) [41], whereas the patterns responded to climate conditions at a large scale (historical contingencies) [42]. The pedogenesis theory might be a possible hub that linking Bass-Becking hypothesis with climate in structuring elevational distribution patterns of soil microbes. According to Dokuchaev, climate is a key determinant of pedogenesis by exerting influences on weathering of rocks, leaching, and biological processes [43]. While biological factors like plant would also impact on the pedogenesis, which determined the soil environmental variable for soil microorganisms. Taken together, both ecological processes and climatic conditions could be very important in determining elevational distribution patterns of soil microbes in the montane ecosystem.
Differential Patterns of Bacterial and Archaeal Taxa
In the second hypothesis, we suggested that soil bacterial and archaeal taxa might distribute differently along the elevational gradients, due to their disparate genetic and metabolic features. In our study, we found that bacterial and archaeal patterns shaped by environmental factors were not exactly the same. The ratio of bacterial to archaeal 16S rRNA gene abundance presented declined variation tendencies along the altitudinal gradient, which demonstrated that bacteria tend to be abundant in the relatively lower altitude while archaea preferred the relatively higher altitude. This result might imply a niche separation of bacteria and archaea at different elevational site, as caused by their differential response to the same environmental factors [44]. The diversity patterns of bacteria and archaea might be more convincing to demonstrate the variations of bacteria and archaea along the vertical altitude gradient. Compared with previous studies which reported a unimodal or declining diversity pattern on merely bacteria [10, 12], our results illustrated that bacterial community rather than archaeal presented a significant decreasing diversity pattern as the altitude increased. The reason might be that diversity of bacterial community was strongly affected by soil pH and clay content which decreased with the increasing elevation [10]. Archaeal community diversity, however, was less influenced by these environmental factors. In addition, we found that altitudinal zonations of bacteria communities were more apparent than archaeal across climate zones. This phenomenon can be supported by that archaea might less participate in the litter decomposition compared with bacteria [45]. The aboveground vegetation is the primary litter contributor to soils, and the highly differences of vegetation across altitudinal climate zones could result in different accumulation of plant residuals. Therefore, microorganisms like bacteria, which are more dependent on the plant litters for substrates, would show more obvious zonation feature than archaea, which are less engaged in the ecological processes of litter decomposition.
Apart from the diversity patterns and elevational zonations of the bacterial and archaeal communities, we also focused on the variations of specific taxa in the community along the elevational gradient. The dominant taxa in bacterial and archaeal community also contributed largely to the differential patterns of bacteria and archaea. For both bacteria and archaea, we found that Acidobacteria in the bacterial communities and MBGA in the archaeal communities were the dominant taxa, which were very sensitive to environmental factors. This finding highlighted the role of soil environmental factors, especially soil pH, in structuring microbial diversity along the elevational gradients. Surprisingly, MBGA, which were usually found to be abundant in marine sediments [46], presented an absolute superiority over other groups in almost all of our samples. They were also detected in another study on soil archaeal communities where only a small portion was observed [45]. We suggested that it might be attributed to a historical contingency of Tibetan Plateau uplifting. As the youngest plateau in the world, the Tibetan Plateau began to rise from the sea since 55 Ma (mega-annum) ago and the formation of southern mountains to the plateau was resulted from the most recent abrupt uplift (<7 Ma) [47]. Considering the relatively shorter geological history of Mt. Shegyla, we presumed that the current soil archaeal communities might preserve some characteristics of the previous marine archaeal communities. This biogeographic pattern of microbes along the altitude gradient investigated in our study, if could be supported by further research, may better describe a microbial geographic region for Tibetan Plateau.
Conclusion
Through this comparative investigation on the soil bacteria and archaea along the elevational gradients of Mt. Shegyla, we observed clear but differential distribution patterns of these two kinds of prokaryotes. Our findings strongly supported that soil prokaryotic microorganisms exhibited community zonations and decreasing diversity patterns along the altitude gradient as macroorganisms. Considering that the soil sampling sites along the elevation are not in a continuous gradient, the distribution patterns of bacteria and archaea merit further study and verification. Our study also highlighted the critical roles of environmental factors in structuring microbial community biogeographic patterns, and might provide new insights into the Bass-Becking hypothesis.
References
Körner C (2007) The use of ‘altitude’ in ecological research. Trends Ecol Evol 22:569–574
Gaston KJ (2000) Global patterns in biodiversity. Nature 405:220–227
Parmesan C, Gaines S, Gonzalez L, Kaufman DM, Kingsolver J, Peterson AT, Sagarin R (2005) Empirical perspectives on species borders: from traditional biogeography to global change. Oikos 108:58–75
Kessler M (2000) Altitudinal zonation of Andean cryptogam communities. J Biogeogr 27:275–282
Romdal TS, Rahbek C (2009) Elevational zonation of afrotropical forest bird communities along a homogeneous forest gradient. J Biogeogr 36:327–336
Fierer N, Jackson RB (2005) The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci U S A 103:626–631
Alexander JM, Kueffer C, Daehler CC, Edwards PJ, Pauchard AB, Seipel T, MIREN Consortium (2010) Assembly of nonnative floras along elevational gradients explained by directional ecological filtering. Proc Natl Acad Sci U S A 108:656–661
Kozak KH, Wiens JJ (2010) Niche conservatism drives elevational diversity patterns in Appalachian salamanders. Amer Nat 176:40–54
Qiong L, Grytnes JA, Birks HJB (2010) Alpine vegetation and species-richness patterns along two altitudinal gradients in the Gyama Valley, south-central Tibet, China. Plant Ecol Divers 3:3,235–247
Bryant JA, Lamanna C, Morlon H, Kerkhoff AJ, Enquist BJ, Green JL (2008) Microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proc Natl Acad Sci U S A 105:11505–11511
Fierer N, McCain CM, Meir P, Zimmermann M, Rapp JM, Silman MR, Knight R (2011) Microbes do not follow the elevational diversity patterns of plants and animals. Ecology 92:797–804
Singh D, Takahashi K, Kim M, Chun J, Adams JM (2012) A hump-backed trend in bacterial diversity with elevation on Mount Fuji, Japan. Microbial Ecol 63:429–437
Colwell RK, Lees DC (2000) The mid-domain effect: geometric constraints on the geography of species richness. Trends Ecol Evol 15:70–76
Yang J, Smith HG, Sherratt TN, Wilkinson DM (2010) Is there a size limit for cosmopolitan distribution in free-living microorganisms? A biogeographical analysis of testate amoebae from polar areas. Microbial Ecol 59:635–645
Wang JJ, Soininen J, Zhang Y, Wang BX, Yang XD, Shen J (2011) Contrasting patterns in elevational diversity between microorganisms and macroorganisms. J Biogeogr 38:595–603
Zhang LM, Wang M, Prosser JI, Zheng YM, He JZ (2009) Altitude ammonia-oxidizing bacteria and archaea in soils of Mount Everest. FEMS Microbiol Ecol 70:208–217
Hu HW, Zhang LM, Dai Y, Di HJ, He JZ (2013) pH-dependent distribution of soil ammonia oxidizers across a large geographical scale as revealed by high-throughput pyrosequencing. J Soil Sediment 13:1439–1449
He JZ, Hu HW, Zhang LM (2012) Current insights into the autotrophic thaumarchaeal ammonia oxidation in acidic soils. Soil Biol Biochem 55:146–154
Wu Z, Tang Y, Li X, Wu S, Li H (1981) Dissertations upon the origin, development and regionalization of Xizang flora through the floristic analysis. Proc Symp Qinghai-Xizang Plat 2:1219–1244
Guo P, Liu Q, Li C, Chen X, Jiang K, Wang YZ, Malhotra A (2011) Molecular phylogeography of Jerdon's pitviper (Protobothrops jerdonii): importance of the uplift of the Tibetan Plateau. J Biogeogr 38:2326–2336
Araújo MB, Whittaker RJ, Ladle RJ, Erhard M (2005) Reducing uncertainty in projections of extinction risk from climate change. Glob Ecol Biogeogr 14:529–538
Prosser JI, Bohannan BJM, Curtis TP, Ellis RJ, Firestone MK, Freckleton RP, Green JL, Green LE, Killham K, Lennon JJ, Osborn AM, Solan M, van der Gast CJ, Young JPW (2007) The role of ecological theory in microbial ecology. Nat Rev Microbiol 5:384–392
Cao P, Zhang LM, Shen JP, Zheng YM, Di HJ, He JZ (2012) Distribution and diversity of archaeal communities in selected Chinese soils. FEMS Microbiol Ecol 80:146–158
Suzuki MT, Taylor LT, DeLong EF (2000) Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl Environ Microbiol 66:4605–4614
Kemnitz D, Kolb S, Conrad R (2005) Phenotypic characterization of Rice Cluster III archaea without prior isolation by applying quantitative polymerase chain reaction to an enrichment culture. Environ Microbiol 7:553–565
Lane D (1991) 16S/23S rRNA sequencing. Wiley, New York
Caporaso JG, Kuczynski J, Stombaugh J et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R (2010) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Faith DP (1992) Conservation evaluation and phylogenetic diversity. Biol Cons 61:1–10
Lozupone CA, Hamady M, Kelley ST, Knight R (2007) Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73:1576–1585
R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/
Oksanen J, Kindt R, Legendre P, O’Hara B (2007) vegan: Community Ecology Package. R package version 2.0-6.Available at: http://cran.r-project.org/
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46
O'Malley MA (2007) The nineteenth century roots of ‘everything is everywhere’. Nat Rev Microbiol 5:647–651
Shen C, Xiong J, Zhang H, Feng Y, Lin X, Li X, Liang W, Chu H (2012) Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol Biochem 54:204–211
Yang Y, Gao Y, Wang S, Xu D, Yu H, Wu L, Lin Q, Hu Y, Li X, He Z, Deng Y, Zhou J (2013) The microbial gene diversity along an elevation gradient of the Tibetan grassland. ISME J 7:1–11
Kourtev PS, Ehrenfeld JG, Haggblom M (2003) Experimental analysis of the effect of exotic and native plant species on the structure and function of soil microbial communities. Soil Biol Biochem 35:895–905
Zheng YM, Cao P, Fu B, Hughes JM, He JZ (2013) Ecological drivers of biogeographic patterns of soil archaeal community. PLoS One 8:e63375
Kozak KH, Wiens JJ (2007) Climatic zonation drives latitudinal variation in speciation mechanisms. P Roy Soc B-Biol Sci 274:2995–3003
Jenny H (1941) Factors of soil formation: a system of quantitative pedology. McGraw-Hill, New York
Adair KL, Schwartz E (2008) Evidence that ammonia-oxidizing archaea are more abundant than ammonia-oxidizing bacteria in semiarid soils of northern Arizona, USA. Microbial Ecol 56:420–426
Singh D, Takahashi K, Adams JM (2013) Elevational patterns in archaeal diversity on Mt. Fuji PLoS One 7:e44494
Inagaki F, Nunoura T, Nakagawa S, Teske A, Lever M, Lauer A, Suzuki M, Takai K, Delwiche M, Colwell FS (2006) Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc Natl Acad Sci U S A 103:2815–2820
Wang CS, Zhao XX, Liu ZF, Lippert PC, Graham SA, Coe RS, Yi HS, Zhu LD, Liu S, Li YL (2008) Constraints on the early uplift history of the Tibetan Plateau. Proc Natl Acad Sci U S A 105:4987–4992
Acknowledgments
This work was financially supported by grants from National Science Foundation of China (41230857, 41025004), MOST (2013CB956300), and STSN-21-02. We gratefully acknowledge Drs Mu Wang and Xi Zha from Agricultural and Animal Husbandry College of Tibet for their assistance in soil sampling.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Spearman’s correlation analysis on environmental variable and diversity index of Shannon (a) and Faith PD (b) (DOC 98 kb)
Rights and permissions
About this article

Cite this article
Wang, JT., Cao, P., Hu, HW. et al. Altitudinal Distribution Patterns of Soil Bacterial and Archaeal Communities Along Mt. Shegyla on the Tibetan Plateau. Microb Ecol 69, 135–145 (2015). https://doi.org/10.1007/s00248-014-0465-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-014-0465-7