
Abstract
In soils, bacteria are very abundant and diverse. They are involved in various agro-ecosystem processes such as the nitrogen cycle, organic matter degradation, and soil formation. Yet, little is known about the distribution and composition of bacterial communities through the soil profile, particularly in agricultural soils, as most studies have focused only on topsoils or forest and grassland soils. In the present work, we have used bar-coded pyrosequencing analysis of the V3 region of the 16S rRNA gene to analyze bacterial diversity in a profile (depths 10, 25, and 45 cm) of a well-characterized field of winter wheat. Taxonomic assignment was carried out with the Ribosomal Database Project (RDP) Classifier program with three bootstrap scores: a main run at 0.80, a confirmation run at 0.99, and a run at 0 to gain information on the unknown bacteria. Our results show that biomass and bacterial quantity and diversity decreased greatly with depth. Depth also had an impact, in terms of relative sequence abundance, on 81 % of the most represented taxonomic ranks, notably the ranks Proteobacteria, Bacteroidetes, Actinobacteridae, and Acidobacteria. Bacterial community composition differed more strongly between the topsoil (10 and 25 cm) and subsoil (45 cm) than between levels in the topsoil, mainly because of shifts in the carbon, nitrogen, and potassium contents. The subsoil also contained more unknown bacteria, 53.96 % on the average, than did the topsoil, with 42.06 % at 10 cm and 45.59 % at 25 cm. Most of these unknown bacteria seem to belong to Deltaproteobacteria, Actinobacteria, Rhizobiales, and Acidobacteria.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Bacteria are the most abundant and diverse microorganisms in soils. They play an important role in soil formation, contribute to plant nutrition, and are involved in various ecosystem functions, such as nutrient cycling, but many things remain to discover about bacterial soil communities: how environmental factors affect them and how their alteration affects ecosystems. This knowledge gap is due principally to the incredible diversity and complexity of the soil [1, 2]. Of the more than 50 bacterial phyla believed to exist, at least 32 are thought to be present in soils [3], and nearly half of these would appear to comprise only bacteria that have never been cultured [4, 5]. Furthermore, many studies based on soil bacterial communities have focused on forest soil [6–8] or grassland soil [9, 10], and although an increasing number of recent studies have analyzed the bacterial diversity of agricultural soils, these studies have focused generally on the topsoil (0–10 cm) [11, 12] or on mixed soil samples corresponding to depths from 0 to 20 or 30 cm (plowing layer) [13–15]. This is a problem, as Fierer et al. in 2003 [16] and Schutz et al. [17] have respectively found 35 and 50 % of the microbial biomass to be present in the subsoil (below 25 cm in the former study and below 40 cm in the latter). Furthermore, most wheat roots extend through the 0–60-cm horizon [18].
Until recently, bacterial diversity was analyzed by techniques such as denaturing gradient gel electrophoresis, automated ribosomal intergenic spacer analysis, or terminal restriction fragment length polymorphism analysis. These techniques provide access to only a small fraction of the total bacteria present in an environment. Today, high-throughput sequencing allows large-scale analysis of the microbial composition of an environment [19, 20] and is widely used in bacterial diversity analyses. Taxonomic assignment of sequences is usually carried out with the Ribosomal Database Project (RDP) Classifier program [21], with a bootstrap score of 0.80 to 0.99. This program is based on a naïve Bayesian assignment algorithm and gives bootstrap scores for the assigned ranks. The bootstrap score decreases as the taxonomic rank assignment becomes more specific (from domain to genus). Using a threshold bootstrap score to sort the rank assignments introduces a new dimension into the analysis, as only the best assignments are kept, at the cost of generating a category of unclassified reads for each rank (and this category may be large in a very diversified environment such as a soil).
Here, we have investigated the diversity of bacterial communities at three depths (10, 25, and 45 cm) in a conventional tillage and a simplified tillage plot in a well-characterized field of winter wheat. The depth of 10 cm corresponds with the tilled layer in both tillage systems, 25 cm corresponds with the tilled layer in the conventional tillage plot and with a previously plowed layer in the simplified tillage plot, and 45 cm corresponds with an undisturbed layer in both plots. Our objectives were to demonstrate, in an agricultural soil, the impact of depth on the known and unknown members of bacterial communities and to detect correlations between soil bacterial communities and physicochemical parameters of the soil. For this, we used high-throughput bar-coded pyrosequencing and the RDP Classifier program for taxonomic assignment of the reads. This assignment was done with three bootstrap scores: 0, 0.80, and 0.99. The score 0.80 is recommended by Wang et al., and its accuracy ranges from 83.2 % for the genus to 99.5 % for the phylum. It was used to analyze shifts in bacterial communities between the three depths. The score 0.99 is very stringent and was used to confirm the accuracy of assignments obtained with score 0.80. Finally, the score 0 was used to assign all reads, each read being therefore assigned to its closest known genus. This score is very lax and was used to calculate for each rank the contribution of reads that could not be assigned in the run at score 0.80.
Methods
Soil Description and Sampling
The experimental site is located in Gembloux (50° 33′ N, 4° 42′ E), Belgium. The soil is a luvisoil (FAO classification). For the topsoil (0 to 30 cm), the texture is silt loam (clay 14–16 %; silt 75–80 %; sand 5–6 %) and the pH is neutral (6.5–7.0). For the subsoil (30–100 cm), the texture is silt loam (clay 20–25 %; silt 70–75 %; sand 3–6 %) and the pH is slightly acidic (6.2–6.5). The structure is blocky angular to blocky subangular/compact. A winter wheat crop was grown on the field in 2011 and again in 2012. The field was divided into plots subjected to simplified tillage (surperficial works 0–10 cm depth) or conventional tillage (plowing depth 30 cm). The simplified tillage scheme began in 2007.
Samplings were performed on 18 May 2011 and 30 May 2012 in a conventional tillage and a simplified tillage plot, the same plots both years, at three depths: 10, 25, and 45 cm. For each year, soil samples were taken in triplicates in two places for each plot and depth. The triplicates were then gathered on a composite sample. The following parameters were measured directly on these composite samples: pH, soil moisture, and soil total nitrogen, carbon, potassium, magnesium, and calcium contents.
DNA Extraction
Eight grams of each composite sample was used to extract genomic DNA from the soil with the PowerMax™ Soil DNA Isolation Kit (MO BIO Laboratories, Solana Beach, CA) according to the manufacturer’s instructions. The quality of the DNA was checked by gel electrophoresis, and it was quantified with the Qubit Fluorometer (Invitrogen, Carlsbad, CA, USA) prior to storage at −20 °C. This quantification was used to calculate the biomass which represents the DNA quantity extracted from 1 g of soil.
Quantification of Total Bacteria and 16S rRNA Genes by qPCR
Total bacteria (in Escherichia coli equivalents/g of dry soil) and the total concentration of 16S rDNA (in gene copies/g of dry soil) were determined on each composite sample by real-time PCR amplification of the DNA segment encoding the V3–V4 region of the 16S rDNA. The primers used were All-Bact-F (forward) 5′-TCCTACGGGAGGCAGCAGT-3′ and All-Bact-R (reverse) 5′-GGACTACCAGGGTATCTAATCCTGTT-3′ [22]. The hydrolysis probe was All-Bact-P (6-FAM)-5′-CGTATTACCGCGGCTGCTGGCAC-3′-(TAMRA) [22]. Real-time PCR was carried out on a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with the following cycle conditions: 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. The PCR reaction mixture consisted of 12.5 μl TaqMan® Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA), 100 nM of each primer, 0.625 μl BSA (4 μg/μl), 100 nM probe, and 5 μl of gDNA (5 ng/μl) in a total volume of 25 μl. The standard curve yielding E. coli equivalents was constructed by PCR amplification of the same target gene in gDNA extracted from known numbers of cells from a culture of E. coli XL1 Blue, and the results were converted to a number of 16S rDNAs by multiplying by 7, i.e., the number of 16S rRNA gene copies per E. coli genome.
Pyrosequencing
The hypervariable V3 region of the 16S rRNA gene was amplified by PCR with bar-coded primers 337F (5′-GACTCCTACGGGAGGCWGCAG-3′) and 533R (5′-TTACCGCGGCTGCTGGCAC-3′). The PCR reaction was carried out in a total volume of 50 μl containing 5 μl tenfold reaction buffer, 500 μM of each deoxynucleotide triphosphate, 0.01 % BSA, 200 mM of each primer, 1.5 U AmpliTaq Gold 360 DNA Polymerase (Applied Biosystems, Foster City, CA, USA), and 50 ng genomic DNA of each composite sample. The following PCR program was used: initial denaturation at 95 °C for 11 min, 30 cycles of 95 °C for 30 s, 48 °C for 30 s, and 72 °C for 2 min, followed by a final elongation step of 72 °C for 35 min. After the amplification, the size and specificity of each PCR product were evaluated by gel electrophoresis (1.5 % agarose). The products were pooled in equimolar concentrations, and the pool was purified and concentrated with the High Pure PCR Product Purification Kit (Roche Diagnostics, Basel, Switzerland) according to the manufacturer’s instructions. This product (100 ng to 1 μg) was used for DNA sequencing on the Ion Torrent PGM Platform. The library was built with the Ion Plus Fragment Library Kit (Life Technologies, Foster City, CA, USA) and the Agencourt AMPure XP Kit (Beckman Coulter, Brea, CA, USA). The quality of the library was checked on an Agilent 2100 Bioanalyser with a High Sensitivity DNA chip (Agilent Technologies, Palo Alto, CA, USA) and quantified with the Ion Library Quantification Kit (Life Technologies, Foster City, CA, USA). Clonal amplification of the library was done on a OneTouch instrument with the Ion OneTouch 200 Reagents Kit, and template preparation for the sequencing reaction was performed with the Ion OneTouch ES instrument. The template was loaded onto an Ion 316 chip and sequenced on the Ion PGM System with the Ion PGM 200 sequencing kit.
Sequence Analysis
The raw reads obtained from the high-throughput sequencing step were processed through two different filters in order to retain only the reads with the highest quality, i.e., reads with a low rate of sequencing error. Firstly, reads lacking a valid pool tag sequence and/or a valid primer sequence were discarded. The tag and primer sequences were then removed from the reads and, on the basis of the tag sequence, each read was assigned to the corresponding sample. Secondly, reads shorter than 50 bp were eliminated from the analysis remaining to a total of 1,598,114 quality reads. After the filtering step, the reads were assigned with the RDP Classifier program with bootstrap scores 0, 0.80, and 0.98 as described in the “Introduction” and the related publications. The number of sequences corresponding to each identified rank was divided by the total number of sequences retained after filtering and multiplied by 100 to yield a relative abundance expressed as a percentage. The Shannon index was used to estimate bacterial diversity, as recommended by Hill et al. [23].
Statistical Analysis
All physicochemical and biological parameters were log2-transformed before statistical analysis. The effect of depth on the physicochemical and biological parameters (biomass and Shannon diversity index) was assessed using ANOVA with the R software. Relationships between bacterial communities and physicochemical parameters were determined with the Spearman rank correlations (XLSTAT 2012) and redundancy analyses (RDA, vegan-R package) [24].
Results
Physicochemical and Biological Parameters
Soil samples were taken in May 2011 and May 2012, at different depths, from a plowed and an unplowed plot. The depths were chosen to allow a comparison of topsoil samples (taken at 10 and 25 cm) with deep soil samples (taken at 45 cm). Several physicochemical and biological parameters were measured directly on the samples (Table 1). These parameters proved relatively constant in the topsoil, but some were lower in the deep soil. Both biological parameters measured, the biomass (P value < 0.001) and the bacterial diversity (P value < 0.01) represented by the Shannon diversity index (Table 1), were found to decrease very significantly with increasing depth. Biomass dropped from 9.75 μg DNA/g dry soil at 10 cm to 1.38 μg DNA/g dry soil at 45 cm and the bacterial diversity from 4.60 at 10 cm to 3.98 at 45 cm. The pH (P value < 0.01) and the total nitrogen (P value < 0.001), carbon (P value < 0.001), phosphorous (P value < 0.001), and potassium (P value < 0.001) contents decreased very significantly with depth, while the calcium and magnesium contents were not significantly impacted by depth. The RDA analysis (Fig. 1a) showed clear differences in bacterial communities according to the depth, the difference being considerable between the deep soil and the top soil. This variation appeared associated mainly with the variation of the potassium, carbon, and total nitrogen contents between deep soil and top soil. In addition, these parameters appeared strongly linked. Furthermore, there was no variation in bacterial community composition according to the year or management type.

Redundancy analysis (RDA) of the most abundant bacterial communities (relative abundances ≥1 %) in a profile of a winter wheat field in relation to physicochemical parameters. RDA of samples (a) and bacterial ranks (b). Samples are in red and begin with soil management (ST simplified tillage, CT conventional tillage) followed by depth (10, 25, or 45 cm) and year (2011 or 2012). Bacterial ranks are in gray and physicochemical parameters in green
Effect of Depth on the Total Bacteria and the Concentration of 16S rRNA Gene Copies
The related quantities of total bacteria (expressed in E. coli equivalents/g of dry soil) and total 16S rDNA (expressed in gene copies/g of dry soil) were determined from the same real-time PCR data (see “Methods”). These quantities were found to decrease slightly (1.3-fold on average) between 10 and 25 cm and greatly (12.5-fold on average) between 25 and 45 cm (Fig. 2). Furthermore, they were, on average, twice as high in 2011 as in 2012. This difference is probably due to the fact that the 2 weeks preceding sampling were dry in 2011 (1.8 mm rainfall) and relatively wet in 2012 (28.2 mm rainfall). This is reflected in the soil humidity measurements (Table 1).

Number of 16S rRNA gene copies at the three depths, in 2011 and 2012, in a plowed and an unplowed plot. ST simplified tillage, CT conventional tillage
Bacterial Community Composition and Diversity
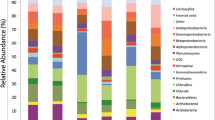
Bacterial diversity was characterized in the various samples by high-throughput sequencing of the hypervariable region V3 of the 16S rRNA gene, followed by sequence assignment with the RDP Classifier program. As the impact of depth was our main focus and the main factor influencing bacterial community composition (Fig. 1a), the results of a first program run (at bootstrap score 0.80, see “Introduction” and “Methods”) were recorded by depth: 10, 25, and 45 cm. This first phylogenetic analysis identified 29 phyla. The communities were found to be composed principally of the following phyla/classes: Alphaproteobacteria, Acidobacteria, Actinobacteria, Bacteroidetes, Nitrospira, Betaproteobacteria, Deltaproteobacteria, Gammaproteobacteria, and Planctomycetes, and the respective average relative abundances of the corresponding sequences were 19.4, 12, 7.5, 3.4, 2.5, 2.1, 2, 1.7, and 0.7 % (Fig. 3). Unknown sequences increased with depth from 42.06 % at 10 cm to 53.96 % at 45 cm, and bacterial diversity, represented by the Shannon index in Table 1, differed more strongly between 25 and 45 cm than between 10 and 25 cm. The sequences corresponding to 38 taxonomic ranks displayed relative abundances above 1 % (Fig. 4 and Supplementary Table 1). Depth had an impact on 81 % of these ranks. This impact was significant (P < 0.05) for 21 % of them, e.g., for Nitrospira, Flavobacteria, and Burkholderiales, and highly significant (P < 0.01) for 60 % of them, e.g., many classes of Proteobacteria, Bacteroidetes, Actinomycetales, and many classes of Acidobacteria. Some Acidobacteria subdivisions, e.g., subdivisions 6 and 4, appeared to decrease with increasing depth, while others, e.g., 1, 3, and 16, appeared to increase. The relative abundance of Bacteroidetes sequences was found to decrease with depth, from 5.84 % at 10 cm to 0.73 % at 45 cm. This decrease was due to the drop in Bacteroidetes incertae-sedis (consisted only of the genus Ohtaekwangia), Chitinophagaceae, and Flavobacteriaceae sequences (Supplementary Table 1). Likewise, Deltaproteobacteria sequences (because of the drop in Myxococcales sequences) were found to decrease from 2.75 % at 10 cm to 0.77 % at 45 cm. Within the class Alphaproteobacteria, depth appeared to affect principally the order Sphingomonadales, whose sequences decreased in relative abundance from 4.89 % at 10 cm to 1.53 % at 45 cm. The relative abundance of Betaproteobacteria and Gammaproteobacteria sequences also varied, from 2.76 and 2.30 %, respectively, at 10 cm to 1.21 % and 0.64 % at 45 cm. Sequences corresponding to the phylum Nitrospira, and essentially Nitrospira sp., increased with depth from 1.89 % at 10 cm to 2.88 % at 45 cm, unlike those corresponding to the order Actinomycetales, which decreased from 4.07 % at 10 cm to 2.50 % at 45 cm. Most of these results were confirmed in a second RDP Classifier run at the more stringent confidence level 0.99 (Fig. 4 and Supplementary Table 1). The only difference was that the relative abundance of Betaproteobacteria and Nitrospira sequences showed little change with depth, in contrast to the shift observed at score 0.80.

Relative distribution of sequences corresponding to the most abundant phyla/classes at 10, 25, and 45 cm. For each taxonomic rank, the relative abundance of 16S rDNA sequences was obtained by dividing the number of sequences corresponding to that rank number by the total number of 16S sequences retained. Sequences were assigned with confidence score 0.80

Heat map of the composition of bacterial communities present in the soil at three depths: 10, 25, and 45 cm. Thirty-eight taxonomic ranks showed a relative abundance above 1 %. Ranks corresponding to family or an upper rank are displayed in the heat map. The scale shows the relative abundances of the corresponding 16S rDNA sequences, calculated on the basis of an RDP Classifier run with the indicated bootstrap score (0.80, 0.99, or 0). Detailed values were available in the Supplementary Table 1. Asterisk: To increase the color resolution, relative abundances of sequences corresponding to the phylum, Proteobacteria (until 45 %) are not shown on the heat map
Assigning Unknown Sequences
To gain some information on the sequences that proved unassignable when bootstrap score 0.80 was applied, the RDP Classifier program was run with bootstrap score 0 and the relative abundance of sequences corresponding to each rank was calculated as after the program run at bootstrap score 0.80 (Fig. 4). This run made it possible to assign each sequence to its closest known genus. Assigning all sequences had an impact on the calculated relative abundances. Some ranks showed an altered impact of depth after the run at bootstrap score 0: the relative abundance of Deltaproteobacteria sequences appeared to increase with depth (from 11.53 % at 10 cm to 18.38 % at 45 cm) instead of decreasing; that of Actinomycetales sequences, instead of decreasing with depth, remained relatively constant from 10 to 25 cm (at 6.49 and 6.06 %, respectively) and increased to 8.11 % at 45 cm; and the relative abundance of Alphaproteobacteria sequences, instead of remaining relatively constant as depth increased, decreased from 24.47 % at 10 cm to 19.67 % at 45 cm. The relative abundances calculated with score 0, compared to those obtained with score 0.80, indicated that most sequences unassignable with score 0.80 were close to the Deltaproteobacteria (8.78 % at 10 cm, 9.66 % at 25 cm, and 17.61 % at 45 cm), Actinobacteria (4.06 % at 10 cm, 4.64 % at 25 cm, and 9.11 % at 45 cm), Rhizobiales (6.89 % at 10 cm, 6.18 % at 25 cm, and 5.22 % at 45 cm), and Acidobacteria (6.71 % at 10 cm, 7.99 % at 25 cm, and 5.27 % at 45 cm). An assignment percentage, i.e., the percentage ratio of sequences assigned with score 0.80 to sequences assigned with score 0, was also calculated (Fig. 5). This assignment percentage varied considerably according to the taxonomic rank. For the well-known Nitrospira phylum, for example, it was 85.55, 88.62, and 95.40 % at 10, 25, and 45 cm, respectively, while for Deltaproteobacteria, it was only 23.84, 19.83, and 4.18 %, and for Acidobacteria, it ranged from 61.61 to 70.06 %.

Heat map of the percentages of assignment. The percentages were calculated for each taxonomic rank by dividing the relative abundance of the relevant sequences obtained after the RDP Classifier run at 0.80 by that obtained after the run at score 0. Detailed values were available in the Supplementary Table 2
Correlations with Physicochemical Parameters
We next examined to what extent the composition of bacterial communities might correlate with pH, soil moisture, or the total nitrogen, carbon, potassium, magnesium, or calcium contents. Spearman correlation coefficients were calculated, and significant correlations were found between bacterial communities and these parameters (Table 2). Soil pH was found to correlate positively with the relative abundance of sequences corresponding to Acidobacteria subdivisions 6 and 4, Bacteroidetes, Actinobacteridae, and Beta/Delta/Gammaproteobacteria. Nitrogen, carbon, and potassium were strongly related (Fig. 1a) and found to correlate positively with the relative abundances of sequences of Bacteroidetes, Beta/Delta/Gammaproteobacteria, Sphingomonadales, Actinobacteridae, and Acidobacteria subdivisions 6 and 4 and negatively with sequences corresponding to Acidobacteria subdivisions 1 and 3. The relative abundances of sequences of Acidobacteria subdivisions 6 and 4, Bacteroidetes, and Beta/Delta/Gammaproteobacteria increased with the concentration of phosphorous. The relative abundance of Actinobacteria sequences was found to correlate only, and negatively, with soil moisture and magnesium, while Nitrospira sequences showed no correlation with any measured parameter.
Finally, RDA analysis was used to illustrate correlations between physicochemical parameters and bacterial communities (Fig. 1). A total of 89 % of the variations observed in bacterial community composition was explained by the first RDA axis. The potassium, carbon, and total nitrogen contents were identified as the most important parameters explaining bacterial community composition. This analysis confirms results obtained with the Spearman correlation (Table 2).
Discussion
On the basis of our high-throughput sequencing data, we have investigated the composition of bacterial soil of a winter wheat field at three depths (10, 25, and 45 cm), in relation to various soil parameters. Overall, our results highlight the relative similarity of soil samples taken at 10 and 25 cm but a real difference in samples taken at 45 cm, at which many physicochemical parameters are much lower than at 10 or 25 cm. This notably concerns the carbon, the potassium, and the nitrogen contents. In keeping with previous studies [6, 16, 25], our data show a strong decrease in biomass and bacterial quantity with increasing depth. This is paralleled by a decrease in bacterial diversity, as also described previously [8, 10, 26, 27]. These effects seem to be mainly due to the decrease in the quantity, and probably the quality [28], of carbon [29].
We have identified 29 phyla in our soil samples. Among the dominant taxonomic ranks, 81 % show an impact of depth. The phyla Proteobacteria and Bacteroidetes, the subclass Actinobacteridae, and many classes of Acidobacteria emerge as the most affected (Fig. 4). The downshift in the relative abundance of Bacteroidetes sequences, due principally to the drop in sequences corresponding to the class B. incertae-sedis and families Chitinophagaceae and Flavobacteriaceae, is the most striking. Such an effect has been reported previously by Will et al. [10] and Eilers et al. [8]. It has been suggested that bacteroidetes are copiotrophic and more abundant in soils containing a lot of organic carbon [29]. We accordingly observe a correlation between the soil carbon content and this phylum (Table 2 and Fig. 1b). In our samples, Ohtaekwangia appears as the most abundant Bacteroidetes genus, representing about 25 % of this phylum. It is a novel genus isolated by Yoon et al. in 2011 [30] from marine sand, and in soils, it has only been referenced so far in the sunflower rhizosphere [31].
In the phylum Acidobacteria, we find, like Janssen [3], subdivisions 1, 4, and 6 to be those most abundantly represented. It is noteworthy that many acidobacterial subdivisions appear to vary greatly between our topsoil (10–25 cm) and deep soil samples (45 cm). Some, such as subdivisions 6 and 4, show a decrease with increasing depth, while others, such as subdivisions 1, 3, and 16, show an increase. In contrast to the results of Jones et al. [32], who found all Acidobacteria subdivisions to correlate (negatively or positively) with soil pH, we find only two subdivisions (4 and 6) to correlate (positively) with this parameter (Table 2 and Fig. 1). This discrepancy is probably due to the fact that only two-thirds of the putative Acidobacteria sequences present in our samples were assigned in the run at score 0.80 (Fig. 5). Accordingly, only members of subdivision 1 are easily isolated and cultured [33–36], whereas only a few members of subdivisions 2, 3, 4, 8, and 10 [35, 37, 38] and none of subdivisions 6 and 7 [3] have been isolated. It is noteworthy, however, that we evidence here a strong positive correlation between subdivisions 4 and 6 and the phosphorus, nitrogen, and carbon contents, a negative correlation between subdivisions 1 and 3 and the nitrogen, carbon, and potassium contents, and a negative correlation between subdivision 16 and the potassium content only.
Alphaproteobacteria are proposed by Fierer et al. [29] to be oligotrophic. We, accordingly, find no correlation between the relative abundance of Alphaproteobacteria sequences and the soil carbon content (Table 2 and Fig. 1). Yet, we do show an overall decrease in Proteobacteria sequences with increasing depth, as observed by Will et al. in a grassland soil [10] and by Eilers et al. in a forest soil [8]. Some proteobacterial taxa are viewed, like the Bacteroidetes, as copiotrophic [7, 29, 39]. This might explain some of the decreases observed, as beta-, delta-, and gammaproteobacterial sequences are all found here to decrease with depth and to show a strong correlation with the carbon content (Table 2 and Fig. 1). Yet, we also evidence positive correlations between these sequences and the nitrogen, potassium, and phosphorus contents. It is worth noting that conclusions regarding the Deltaproteobacteria should be considered with caution because this class is not well-known and only 23.84, 19.83, and 4.18 % of the sequences collected at 10, 25, and 45 cm, respectively, were assigned in the run at score 0.80 (Fig. 5). Furthermore, the results of the run at score 0 contradict the findings of the run at score 0.80 (Fig. 4): they show an increase in deltaproteobacterial sequences with increasing depth.
The reality is likely to be closer to the results obtained with score 0, which includes all the reads obtained by high-throughput sequencing. Our poor knowledge of Deltaproteobacteria is confirmed by Janssen in 2006 and is due to the focus of most studies on unknown Acidobacteria and Verrucomicrobia rather than on unknown Deltaproteobacteria.
Our runs at score 0.80 and score 0 yield conflicting results for Actinomycetales sequences; also, a decrease with depth at score 0.80 and no change in the topsoil followed by an increase at 45 cm at score 0. One should stress that many Actinomycetales genera remain unknown [3], and the percentages of assignment observed here (62.70, 54.51, and 30.83 % at 10, 25, and 45 cm, respectively) suggest that the proportion of unknown Actinobacteridae members increases with increasing depth. This might explain the discrepancy. Unlike the other taxonomic ranks studied here, the Actinobacteria seem not to be influenced by the measured physicochemical parameters, apart from the observed negative correlations with soil moisture and magnesium content. The Actinobacteria are indeed believed to be adapted to resource-limited conditions and to life in the deep soil [40, 41], where competition between bacteria is furthermore less important.
In conclusion, we have investigated the diversity of bacterial communities in a profile of a winter wheat field with a new taxonomic assignment approach. The depth appears to affect most of the bacterial taxonomic ranks identified here, and Proteobacteria, Bacteroidetes, Acidobacteria, and Actinobacteridae appear as the most affected. The observed shifts correlate with shifts in physicochemical parameters, especially the carbon, nitrogen, and/or potassium content, and/or the decrease in oxygen with increasing depth. The observed changes in parameters and communities are more pronounced between 25 and 45 cm than between 10 and 25 cm, and the number of unknown bacteria increases with depth. By including an RDP Classifier run at score 0, in addition to the main run at 0.80 and a confirmation run at 0.99 (yielding good confirmation in most cases), we have been able to gain information on the unknown as well as the known bacteria in our samples. The results obtained with score 0 highlight the bias introduced at higher confidence levels: if one considers the samples taken at 45 cm, for example, the assignment percentage of the run at score 0.80 versus the run at score 0 was only 4.18 % for deltaproteobacterial sequences and 18.81 % for sphingobacterial sequences. The assignment percentage of a run at score 0.95 to 0.98 would have been even lower, and these scores are sometimes used in the literature [42–44]. This bias can lead to erroneous conclusions (see the above-mentioned conflicting results regarding the influence of depth on deltaproteobacteria). We therefore recommend using bootstrap score 0 in addition to a higher score such as 0.50 or 0.80 when assigning sequences with RDP Classifier. Yet, the use of score 0 does not enable one to identify the exact species, and agricultural soils contain many unknown species that remain to be identified and studied.
Sequence Accession Number
Pyrotag sequences reported in this study were deposited in the GenBank database with the accession number SRP029463.
References
Curtis TP, Sloan WT, Scannell JW (2002) Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci U S A 99(16):10494–10499
Gans J, Wolinsky M, Dunbar J (2005) Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309(5739):1387–1390
Janssen PH (2006) Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72(3):1719–1728
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180(18):4765–4774
Rappé MS, Giovannoni SJ (2003) The uncultured microbial majority. Annu Rev Microbiol 57:369–394
Hartmann M, Lee S, Hallam SJ, Mohn WW (2009) Bacterial, archaeal and eukaryal community structures throughout soil horizons of harvested and naturally disturbed forest stands. Environ Microbiol 11(12):3045–3062
Nemergut DR, Cleveland CC, Wieder WR, Washenberger CL, Townsend AR (2010) Plot-scale manipulations of organic matter inputs to soils correlate with shifts in microbial community composition in a lowland tropical rain forest. Soil Biol Biochem 42(12):2153–2160
Eilers KG, Debenport S, Anderson S, Fierer N (2012) Digging deeper to find unique microbial communities: the strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol Biochem 50:58–65
Thomson BC, Ostle N, McNamara N, Bailey MJ, Whiteley AS, Griffiths RI (2010) Vegetation affects the relative abundances of dominant soil bacterial taxa and soil respiration rates in an upland grassland soil. Microb Ecol 59(2):335–343
Will C, Thürmer A, Wollherr A, Nacke H, Herold N, Schrumpf M et al (2010) Horizon-specific bacterial community composition of German grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microbiol 76(20):6751–6759
Preem JK, Truu J, Truu M, Mander Ü, Oopkaup K, Lõhmus K et al (2012) Bacterial community structure and its relationship to soil physico-chemical characteristics in alder stands with different management histories. Ecol Eng 49:10–17
Rohini-Kumar M, Osborne JW, Saravanan VS (2013) Comparison of soil bacterial communities of Pinus patula of Nilgiris, western ghats with other biogeographically distant pine forest clone libraries. Microb Ecol 66(1):132–144
Navarrete AA, Cannavan FS, Taketani RG, Tsai SM (2010) A molecular survey of the diversity of microbial communities in different Amazonian agricultural model systems. Diversity 2:787–809
Orr CH, James A, Leifert C, Cooper JM, Cummings SP (2011) Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Appl Environ Microbiol 77(3):911–919
Ding GC, Piceno YM, Heuer H, Weinert N, Dohrmann AB, Carrillo A et al (2013) Changes of soil bacterial diversity as a consequence of agricultural land use in a semi-arid ecosystem. PLoS ONE 8(3):e59497. doi:10.1371/journal.pone.0059497
Fierer N, Schimel JP, Holden P (2003) Variations in microbial community composition through two soil depth profiles. Soil Biol Biochem 35:167–176
Schutz K, Kandeler E, Nagel P, Scheu S, Ruess L (2010) Functional microbial community response to nutrient pulses by artificial groundwater recharge practice in surface soils and subsoils. FEMS Microbiol Ecol 72:445–455
White RG, Kirkegaard JA (2010) The distribution and abundance of wheat roots in a dense, structured subsoil-implications for water uptake. Plant Cell Environ 33(2):133–148
Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AK, Kent AD, Daroub SH, Camargo FA, Farmerie WG, Triplett EW (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1:283–290
Uroz S, Buée M, Murat C, Frey-Klett P, Martin F (2010) Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ Microbiol Rep 2:281–288
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Nadkarni MA, Martin FE, Jacques NA, Hunter N (2002) Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148:257–266
Hill TC, Walsh KA, Harris JA, Moffett BF (2003) Using ecological diversity measures with bacterial communities. FEMS Microbiol Ecol 43(1):1–11
Oksanen J, Blanchet FG, Kindt R, Legendre P, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H (2011) Vegan: community ecology package. R package version 1.17-7
Ekelund F, Rønn R, Christensen S (2001) Distribution of microbial biomass and phospholipid fatty acids in Podzol profiles under coniferous forest. Soil Biol Biochem 33:475–481
Zhou J, Guo W, Wang R, Han X, Wang Q (2008) Microbial community diversity in the profile of an agricultural soil in northern China. J Environ Sci (China) 20(8):981–988
Goberna M, Insam H, Klammer S, Pascual JA, Sánchez J (2005) Microbial community structure at different depths in disturbed and undisturbed semiarid Mediterranean forest soils. Microb Ecol 50(3):315–326
Rumpel C, Kögel-Knabner I (2011) Deep soil organic matter—a key but poorly understood component of terrestrial C cycle. Plant Soil 338:143–158
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88(6):1354–1364
Yoon JH, Kang SJ, Lee SY, Lee JS, Park S (2011) Ohtaekwangia koreensis gen. nov., sp. nov. and Ohtaekwangia kribbensis sp. nov., isolated from marine sand, deep-branching members of the phylum Bacteroidetes. Int J Syst Evol Microbiol 61:1066–1072
Tejeda-Agredanoa MC, Gallegob S, Vilab J, Grifollb M, Ortega-Calvoa JJ, Cantosa M (2013) Influence of the sunflower rhizosphere on the biodegradation of PAHs in soil. Soil Biol Biochem 57:830–840
Jones R, Robeson M, Lauber C, Hamady M, Knight R, Fierer N (2009) A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J 3:442–453
Kishimoto N, Tano T (1987) Acidophilic heterotrophic bacteria isolated from acidic mine drainage, sewage and soils. J Gen Appl Microbiol 33:11–25
Sait M, Hugenholtz P, Janssen PH (2002) Cultivation of globally distributed soil bacteria from phylogenetic lineages previously only detected in cultivation-independent surveys. Environ Microbiol 4:654–666
Davis KER, Joseph SJ, Janssen PH (2005) Effects of growth medium, inoculum size, and incubation time on the culturability and isolation of soil bacteria. Appl Environ Microbiol 71:826–834
Sait M, Davis KER, Janssen PH (2006) Effect of pH on isolation and distribution of members of subdivision 1 of the phylum Acidobacteria occurring in soil. Appl Environ Microbiol 72:1852–1857
Joseph SJ, Hugenholtz P, Sangwan P, Osborne CA, Janssen PH (2003) Laboratory cultivation of widespread and previously uncultured soil bacteria. Appl Environ Microbiol 69:7210–7215
Foesel BU, Rohde M, Overmann J (2013) Blastocatella fastidiosa gen. nov., sp. nov., isolated from semiarid savanna soil—the first described species of Acidobacteria subdivision 4. Syst Appl Microbiol 36(2):82–89
Eilers G, Lauber CL, Knight R, Fierer N (2010) Shifts in bacterial community structure associated with inputs of low molecular weight carbon compounds to soil. Soil Biol Biochem 42(6):896–903
McCarthy AJ, Williams ST (1992) Actinomycetes as agents of biodegradation in the environment—a review. Gene 115(1–2):189–192
Kirby R (2005) Actinomycetes and lignin degradation. Adv Appl Microbiol 58C:125–168
Webster NS, Taylor MW, Behnam F, Lücker S, Rattei T, Whalan S et al (2010) Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ Microbiol 12(8):2070–2082
Li RW, Connor EE, Li C, Baldwin Vi RL, Sparks ME (2012) Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ Microbiol 14(1):129–139
Giongo A, Favet J, Lapanje A, Gano KA, Kennedy S, Davis-Richardson AG (2013) Microbial hitchhikers on intercontinental dust: high-throughput sequencing to catalogue microbes in small sand samples. Aerobiologia 29(1):71–84
Acknowledgments
We would like to thank Gembloux Agro-Bio Tech - University of Liège for financially supporting this study.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Composition of bacterial communities present in the soil at three depths: 10, 25, and 45 cm. Relative abundances of the corresponding 16S rDNA sequences were calculated on the basis of an RDP Classifier run with the bootstrap scores of 0.80, 0.99, or 0 (XLS 36 kb)
Supplementary Table 2
Percentages of assignment. Percentages were calculated for each taxonomic rank by dividing the relative abundance of the relevant sequences obtained after the RDP Classifier run at 0.80 by that obtained after the run at score 0 (XLS 32 kb)
Rights and permissions
About this article

Cite this article
Stroobants, A., Degrune, F., Olivier, C. et al. Diversity of Bacterial Communities in a Profile of a Winter Wheat Field: Known and Unknown Members. Microb Ecol 68, 822–833 (2014). https://doi.org/10.1007/s00248-014-0458-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-014-0458-6