
Abstract
Caves are relatively accessible subterranean habitats ideal for the study of subsurface microbial dynamics and metabolisms under oligotrophic, non-photosynthetic conditions. A 454-pyrotag analysis of the V6 region of the 16S rRNA gene was used to systematically evaluate the bacterial diversity of ten cave surfaces within Kartchner Caverns, a limestone cave. Results showed an average of 1,994 operational taxonomic units (97 % cutoff) per speleothem and a broad taxonomic diversity that included 21 phyla and 12 candidate phyla. Comparative analysis of speleothems within a single room of the cave revealed three distinct bacterial taxonomic profiles dominated by either Actinobacteria, Proteobacteria, or Acidobacteria. A gradient in observed species richness along the sampling transect revealed that the communities with lower diversity corresponded to those dominated by Actinobacteria while the more diverse communities were those dominated by Proteobacteria. A 16S rRNA gene clone library from one of the Actinobacteria-dominated speleothems identified clones with 99 % identity to chemoautotrophs and previously characterized oligotrophs, providing insights into potential energy dynamics supporting these communities. The robust analysis conducted for this study demonstrated a rich bacterial diversity on speleothem surfaces. Further, it was shown that seemingly comparable speleothems supported divergent phylogenetic profiles suggesting that these communities are very sensitive to subtle variations in nutritional inputs and environmental factors typifying speleothem surfaces in Kartchner Caverns.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Bacteria have been found in every known ecological niche on earth with the microbial diversity of each niche specifically, finely tuned to exploit the environmental conditions found in that particular ecosystem. Analysis of the microbial diversity of unique habitats provides both the opportunity to expand our understanding of global microbial diversity and the potential to gain insights into the energy dynamics of novel population assemblages. Limestone caves provide one such habitat where growth conditions characterized by mild, stable temperatures, high humidity, circumneutral pH, and abundant oxygen are severely constrained by nutrient-limited conditions. The oligotrophic conditions found in caves are a direct result of the lack of photosynthetic activity due to the absence of sunlight. These conditions force bacteria to rely on alternative primary production strategies to supplement the limited supplies of allocthonous organic carbon sourced from the surface. Further, limestone caves generally lack exogenous energy sources such as those present in caves with sulfide- [1, 2] and ferromanganese-rich deposits [3, 4].
In this study, we focus on the bacterial diversity of Kartchner Caverns, a limestone cave located in the Sonoran Desert of southeastern Arizona. Despite its desert location, humidity within Kartchner Caverns is on average 99.4 % and the cave contains a wide diversity of actively growing calcite speleothems [5]. The oligotrophic conditions typical of caves are further intensified in Kartchner Caverns due to the low amounts of organic matter present in the semiarid soils of the region which impact the quantity of organic carbon reaching cave microbes through drip water. The organic carbon concentration in drip water fluctuates from 0.79 to 2.7 mg C L−1 on an annual basis (Legatzki, unpublished); values similar to caves in other desert regions [6], but three orders of magnitude lower than levels found in the drip water of caves in temperate regions [6, 7].
Previous surveys of microbial community structure conducted on different speleothems in two distinct areas of Kartchner Caverns using PCR-denaturing gradient gel electrophoresis (PCR-DGGE) indicated that the formations support complex communities and that the structure of these communities varied between speleothems located within the same room of the cave [8, 9]. Further, observed differences in community structure on adjacent speleothems were preserved when sampled over two consecutive years [9]. Canonical correspondence analysis of ten actively growing speleothems located within one room of the cave revealed that the variations in bacterial community structure could not be correlated with either physical (dimensions, color) or chemical (elemental and organic carbon concentration of speleothem surface material) properties of the individual formations, but were correlated with the specific location of that formation within the room. These results suggested an association between the observed drip water sources feeding individual stalactites and the respective community structure [8].
The goal of the current study was to further characterize the specific differences in community structure documented by our previous study. The depth of sequencing provided by 454-pyrotag analysis was utilized to accomplish a twofold objective; (1) to quantify the comparative species richness and diversity of the bacterial community groups associated with the speleothems analyzed in the previous study, and (2) to compare the taxonomic composition of each community sampled to identify the magnitude of hierarchical diversity differentiating these distinct communities. In addition, phylogenetic analysis of nearly full-length 16S rRNA genes from one speleothem was used to obtain more specific identifications and to characterize the novelty of the bacterial populations colonizing the calcite speleothems of this limestone cave ecosystem. While previous studies have documented distinct bacterial communities present in limestone caves located in globally diverse regions [8–12], the current work is unique in its effort to document the magnitude of variability in bacterial diversity and taxonomic composition of communities colonizing adjacent speleothems within a single cave. In depth taxonomic characterization of the distinct bacterial communities present in Kartchner Caverns is a critical step for elucidating the functional dynamics driving the formation of bacterial assemblages colonizing calcite speleothems.
Methods
Site Description and Sampling
Kartchner Caverns is a limestone cave characterized by high humidity (99.4 %), CO2 levels that range from 1,000 to 5,000 ppm and a mean annual temperature of 19.8 °C. The only natural entrance to the cave is a single blowhole and so air exchange with the surface is minimal [13]. A Myotis velifer bat population roosts in the cave from late April to mid September and is limited to the Big Room region of the cave [14]. Most other fauna in the cave are arthropods that depend on the guano provided by the bat population [15].
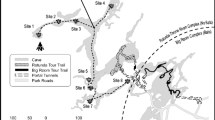
The Big Wall room (BW) located in the Rotunda-Throne room region of Kartchner Caverns was selected as the primary sampling site for this study. Six actively dripping stalactites (A, B, D, E, F, and W) and one bacon drapery (C) (Fig. 1) were sampled in November 2008 by swabbing the surface with sterile swabs (35 cm2 surface area per sample), wet with sterile dH2O. Each formation was sampled with ten swabs that were stored immersed in 6 ml sterile dH2O at 4 °C and processed within 24 h. In addition, a composite sample was collected (10 swabs total) from the surface of two cave rock walls immediately adjacent to the speleothems (Fig. 1) and processed similarly. Extracted community DNA from samples previously collected in April 2007 by Legatzki et al. [9], from two speleothems located in the Strawberry Alcove room (SA) of the Big Room region of the cave, was also included in the analysis (Fig. 1), as a basis of comparison for the BW room samples. The labels used in the previous study for these two samples have been changed from C and S to CA and ST, respectively, to avoid confusion with BW room sample designations. These two rooms of the cave were selected based on a high level of speleothem diversity, low human impact (limited to scientists and rangers, ∼10 people per year) and their location in two distinct regions of the cave. Detailed physical and chemical characterizations of the rooms and formations sampled in this study were described previously in Legatzki et al. [8, 9]. Finally, a soil sample was collected above the cave from the area above the Big Wall room for comparison of soil bacterial diversity to that found within the cave. The soil sample was collected using sterile tools as described in Drees et al. [16]. Briefly, a 100-g soil sample was collected as far as possible from plants to avoid rhizosphere effects and at a depth of 20 cm in order to obtain a sample minimally impacted by eolian dispersion. The sample was placed into sterile polycarbonate tubes and stored on ice for transport back to the laboratory. Soil was then air-dried, sieved with a sterile 2-mm sieve to remove rock debris and stored at −20 °C until processed.

Map of Kartchner Caverns State Park. The cave map indicates the tour trails (Rotunda-Throne and Big Room tour trails), the Big Wall room (BW) and the Strawberry Alcove room (SA). Inset Map of the BW room showing the horizontal distances in m between the speleothems sampled in this area. The speleothems (filled circle) are marked by their alphabetical label. The rock wall sample was a composite from two rock walls (filled square), labelled in the figure as Rock 1 and Rock 2 . The two stalactites sampled in the SA room (CA and ST) were separated by a distance of 2.4 m (not shown)
DNA Extraction
The BW room swab samples were sonicated for 20 s in the 6 ml storage solution, vortexed for 1 min, then sonicated again for 20 s after which the swabs were aseptically removed. Genomic DNA was then extracted using the procedure described by Legatzki et al. [9] for the SA room samples collected in 2007. Potential contamination during the sampling or extraction procedure was addressed by opening sterile swabs in the cave, holding them in the air for 1 min, placing them in sterile dH2O and processing them in parallel with the environmental samples. Soil community DNA was extracted from 0.5 g of soil sample using the FastDNA spin kit for soils (MP Biomedicals, Solon, OH, USA) following the manufacturer’s protocol optimized to enhance DNA recovery from desert soils [17].
Amplicon Library Preparation for 454-pyrosequencing
Amplicon libraries of the 16S rRNA V6 region were generated using a modified version of the primers 967f (5′-MWA CGC GAR GAA CCT TAC C-3′) and 1070r (5′-AGC TGA CGA CAR CCA T-3′) [18] that contained the 454 adaptor primers A or B, respectively. The forward primer included a ten nucleotide barcode sequence unique to each sample. The 25 μl PCR reaction mix contained: 1× Invitrogen High Fidelity PCR Buffer (Invitrogen Corporation, Carlsbad, CA, USA), 2.5 mM of MgSO4, 0.5 μM of each primer, 0.4 g L−1 unacetylated bovine serum albumin (Sigma, St. Louis, MO, USA), 200 μM of each deoxynucleoside triphosphate, 5 % dimethyl sulfoxide, 1.0 U Platinum Taq High Fidelity DNA polymerase (Invitrogen Corporation, Carlsbad, CA, USA) and 100 pg template DNA. Amplification conditions were 94 °C for 2 min; 30 cycles of 94 °C for 30 s, 55 °C for 40 s, and 68 °C for 40 s and a final extension at 68 °C for 5 min. Amplicons from three reactions for each sample were excised from gels, pooled and purified using QIAquick Gel Extraction Kit (Qiagen Inc., Valencia, CA, USA). The samples were submitted to the Arizona Genomic Institute (University of Arizona, Tucson, AZ, USA) for unidirectional sequencing in the forward direction by the 454/Roche GS-FLX-Titanium system. Sequences were deposited in the NCBI Sequence Read Archive under accession number SRA037637.1.
Sequence Analysis and Community Comparison
A total of 551,526 reads were obtained and were screened through the following filters: minimum average quality score of 30, no N’s in the sequence, maximum length 120 bp, minimum length 95 bp and presence of forward and reverse primer sequences. The sequences remaining after filtering (79 %) were trimmed to 75 bp after removal of the forward primer.
Initial operational taxonomic unit (OTU) clustering was carried out using a combination of the software packages Mothur v1.12.2 [19] and ESPRIT [20] with a 0.03 cutoff. A total of 636 OTUs were identified as contaminants based on their presence in high abundance in the blank (i.e. >40 % of the total reads belonging to each of these OTUs were from the blank) processed in parallel throughout this study. Sequences belonging to these contaminant OTUs were removed, the pipeline was re-started with the remaining sequences, and the OTU clustering step re-run. This data set was used for all further analyses. Richness and diversity indices, rarefaction curves and pairwise community comparisons generated using the Sørensen abundance (Sorabund) analysis were computed by the respective Mothur applications (www.mothur.org). Taxonomic classification of the reads was done using the Global Alignment for Sequence Taxonomy (GAST) [21] with an 80 % similarity cutoff. The GAST scripts and database files were obtained from vamps.mbl.edu. This classification method was selected given the high accuracy of its taxonomic assignments when using the V6 hypervariable region as demonstrated by Huse et al. [22]. The most abundant phylotypes were further classified based on a combination of GAST, the Ribosomal Database Project’s Classifier [23] and BLAST analysis against the NCBI database [24]. This step was done to classify dominant OTUs that were unclassified using GAST alone and/or to obtain more detailed taxonomic information when possible. Finally, phylum abundances were calculated based on the total number of sequence reads remaining following the removal of contaminant OTU sequences. DNA sequences for the most abundant phylotypes are provided in the electronic supplementary material (Supplementary Table 1). The complete analysis described above was repeated with data normalized for sample sequence read number, but no significant differences were observed, thus the entire dataset was retained. Results for all the analyses performed can be found at: http://www.agcol.arizona.edu/kartchner/.
Bacterial 16S rRNA Gene Library
Genomic DNA from speleothem D was amplified for preparation of a 16S rRNA gene library using primers 27f and 1492r [25]. The 25 μl PCR reaction mix contained: 1× PCR Buffer C (10× buffer C = 100 mM Tris–HCl, 500 mM KCl, 20 mM MgCl2, pH 8.28), 0.5 μM of each primer, 0.4 g L−1 unacetylated bovine serum albumin (Sigma, St. Louis, MO, USA), 200 μM of each deoxynucleoside triphosphate, 5 % dimethyl sulfoxide, 0.625 U Taq DNA polymerase (Roche Diagnostics GmbH, Mannheim, Germany) and 138–209 pg template DNA. Amplification conditions were 95 °C for 3 min; 30 cycles of 95 °C for 40 s, 55 °C for 40 s, and 72 °C for 84 s and a final extension at 72 °C for 10 min. PCR products from three separate reactions were pooled and purified using the QIAquick PCR Purification Kit (Qiagen Inc., Valencia, CA, USA). Amplicons were cloned into pCR®2.1-TOPO® vector (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. A total of 312 clones were processed by plasmid extraction (Plasmid preparation kit, Fermentas Inc., Glen Burnie, MD, USA) and Sanger-sequenced with primer 518f [25] by the University of Arizona Genetics Core (UAGC, Tucson, AZ, USA). OTUs were generated from the resulting sequence reads based on 99 % similarity using Mothur software [19] and a representative sequence from each OTU group was selected for full-length sequencing using the UAGC vector primers T7f and M13r. Sequences generated from each OTU by the three primers (T7f, 518f, and M13r) were assembled using the Faktory software (University of Arizona, Tucson, AZ) generating nearly full-length 16S rRNA gene sequences. Chimeras were identified with Bellerophon v3 and removed [26]. The full-length sequences were clustered into 57 OTUs at 97 % similarity by Mothur. Representative sequences from each OTU were aligned using the SINA online aligner available through the SILVA online resource [27]. ARB software [28] and BLAST searches [24] were used to identify putative phylogenetic groupings for each OTU. Associated reference sequences were identified with the ARB software and retrieved from SILVA [27] and NCBI databases. Rooted most parsimonious trees were constructed from clone and reference sequences by heuristic search (tree bisection reconnection branch swapping) using PAUP version 4.0 Beta 10 software [29]. The 16S rRNA gene sequences were deposited in the GenBank database under accession numbers JN178885–JN178941.
Results
454-pyrotag Diversity Analysis of Bacterial Communities
The 454-pyrosequencing of eleven samples generated a total of 382,319 sequence reads after quality filtering and contaminant removal, representing 69 % of the original dataset. Sample library size ranged from 23,486 to 46,186 sequence reads. The samples evaluated included seven speleothems and a cave rock wall from the BW room sampled in 2008, two speleothems from the SA room sampled in 2007 and a soil sample from directly above the cave collected in 2008. Diversity indices varied significantly among the sample sites (95 % confidence interval) (Table 1). Within the BW room, observed species richness ranged from 1,568 to 2,422 OTUs with the lowest values being found on speleothems W, D, and E located at one end of the room and the highest values associated with speleothems A and B at the other end of the sampling transect (Table 1, Supplementary Fig. 1). The rock wall species richness was 2,326, comparable to speleothems A and B. The richness of the SA stalactites (1,982–2,241) fell within the range observed for the seven BW formations. Finally, the soil community had an observed richness of 4,533 OTUs, approximately twice that of the cave samples. Percent coverage for these communities based on the ACE estimates was varied (Table 1), ranging from 47 % for speleothem A to 71 % for speleothem W in the BW room. Coverage for the SA room (59–70 %) was within the range observed for the BW room. Coverage was greatest for the soil community (78 %) despite the fact that this community had the highest species richness (Table 1).
The range in the Shannon diversity index for the BW room was 4.32 (Speleothem W) to 6.06 (Speleothem B). Once again, speleothems A and B and the rock wall community were most diverse and speleothems C and W least diverse (Table 1). The average diversity for the SA room (5.17) was similar to the average for the BW room (5.15). Abundant OTUs (≥10 reads) comprised less than 23 % of the bacterial communities for each sample (13.5–22.7 %) while low abundance OTUs (≤2 reads) represented an average of 57 ± 4.2 % of the respective communities. Unique OTU abundance (OTUs specific to a single sample) in each community was found to be 18–23 % for the BW speleothems, 27 % for the BW rock, 28 and 36 % for the two SA speleothems, and 84 % for the soil community. Thus, species overlap was greater among the BW speleothems, than between these speleothems and the adjacent rock wall. Further, just 16 % of the soil OTUs were found in any of the communities sampled from within the cave, suggesting that the cave harbors unique microbial communities that developed independently from the soil bacteria potentially entering the cave in drip water from the surface.
Pairwise community comparisons were computed within the BW and SA rooms using the Sorabund similarity index (Fig. 2). The two community pairs with the greatest similarity were speleothems A/B and C/E. Speleothems A and B are two immediately adjacent, morphologically similar stalactites descending from the same drapery and characterized by the highest level of diversity among the speleothem communities sampled. In contrast, C and E are morphologically distinct formations located 4.2 m apart and physically separated by D, F, and W (Fig. 1). C is a drapery while E is a stalactite. The C and E communities were among the least diverse of those sampled from within the cave (Table 1). Stalactites F and W were the least similar pair of the BW room formations (Fig. 2), though they were located just 0.8 m apart. As would be expected, speleothems CA and ST in the SA room were more similar to each other than to any of the communities analyzed in the BW room (Fig. 2), confirming previously published conclusions based on PCR-DGGE analysis [8].

Pairwise community comparisons. The bar scale represents the Sorabund index, a value between 0 and 1 that compares the bacterial communities between two samples. Darker color squares represent less similarity between sample communities
Analysis of Community Taxonomic Composition
Community taxonomic profiles were generated for each of the samples based on analysis of the 454-pyrotag sequences. Based on these profiles the magnitude of the taxonomic differences driving observed variations in community diversity was evaluated. A total of 21 phyla and 12 candidate phyla were identified from the cave communities although >98 % of the sequences belonged to just 13 phyla and 4 candidate phyla (Fig. 3a). The taxonomic analysis revealed that the speleothems in the BW room separated into three quite distinct community types; Type 1, included speleothems C, D, E, and W; Type 2, included speleothems A and B; and Type 3, was limited to speleothem F.

Bacterial community composition of 454 sequence libraries. a Distribution of dominant phyla in cave and soil samples. The category “others” represents bacterial phyla that were represented in less than 1 % in all samples and include the phyla Spirochaetes, Deinococcus-Thermus, Deferibacteres, Aquificae, Fusobacteria, Lentisphaerae, Chlamydiae, Chlorobi, BRC1, OPB7, OP3, OP5, OP10, OP11, TG1, and OD1. Phylotypes that could not be classified by GAST with more than 80 % similarity to reference sequences were determined as unclassified. b Distribution of Actinobacteria (order) in 454 samples. The orders Coriobacteriales and Bifidobacteriales were only found in formations E and ST, and formation CA, respectively, and represented ≤0.1 % (not shown). c Distribution of Proteobacteria (class) in 454 samples. The class Epsilonproteobacteria was found in all samples except in CA and Soil but represented ≤0.28 % of the sequences (not shown)
Type 1 bacterial communities (C/D/E/W) were dominated by Actinobacteria with abundances ranging from 48 to 66 % (Fig. 3a). The majority of these Actinobacteria belonged to the Actinomycetales followed next by the Rubrobacterales order, and finally by low abundances of Acidimicrobiales (Fig. 3b). The next most abundant phylum in these communities was Proteobacteria. The OTUs within this phylum were fairly evenly distributed between Alphaproteobacteria and Gammaproteobacteria with the former being slightly more abundant (Fig. 3c). Speleothem W deviated slightly from the pattern with 18 % Alphaproteobacteria and just 3 % Gammaproteobacteria. The remaining phyla present in the C/D/E/W communities had abundances <5 % with one exception; 9 % of the speleothem C community belonged to a single OTU in the candidate phylum SBR1093. This anomaly is particularly intriguing because the remaining ten communities evaluated (including the soil) contained abundances <0.2 % for this particular group. No bacteria have been cultured from this candidate phylum and the previously identified clones were from geographically diverse environments; one from an activated sludge sample [30], eight from hydrocarbon contaminated soils in France [31] and one from stromatolites in Shark Bay, Western Australia [32].
Type 2 communities (A and B) were dominated by Proteobacteria with abundances of 40 % for A and 33 % for B (Fig. 3a). The abundance of Actinobacteria was just 32 and 28 % for A and B, respectively, levels that were much lower than those found in the C/D/E/W group. Further, within the Proteobacteria, Deltaproteobacteria abundance was 12 and 5 times higher in the A and B communities, respectively, than the average for the C/D/E/W group. The average abundance of Alphaproteobacteria in the Type 2 communities was actually lower than in Type1, whereas the Gammaproteobacteria averages were comparable for both communities (Fig. 3c). The Type 2 community was also characterized by greater abundances of both Chloroflexi and Planctomycetes than found in the C/D/E/W communities. The average Chloroflexi abundance in the A/B communities was 3.3-fold greater than speleothem D and 15- to 100-fold greater than speleothems C, E, and W. Likewise, Planctomycetes were 5 to14 times higher in A/B than in C/D/E/W.
Finally, the Type 3 community represented by speleothem F was unique among all the speleothems analyzed in both the BW and SA rooms (Fig. 3a). The F community was dominated by Acidobacteria (33 % abundance), a phylum with a maximum abundance of 11 % in all the other BW communities. The abundance of Proteobacteria was just 14 %, the lowest value among all cave communities evaluated. In addition, the speleothem F community included 10 % Gemmatimonadetes and 4.8 % Nitrospira, two phyla present at <4 and <2.2 %, respectively, on the remaining BW speleothems. The taxonomic composition of F is clearly unique among the BW communities, despite the fact that it is located within 1.5 m of D, W, and E (Fig. 1).
The BW rock wall community was comprised of Actinobacteria (41 %), Proteobacteria (37 %), and Acidobacteria (6 %) at abundance levels that were intermediate to those found in the seven BW speleothem communities (Fig. 3a). Unique distributions were only observed for Gammaproteobacteria (20 %) and Firmicutes (1.4 %) where abundances were 1.5- to 2-fold greater than in any of the BW speleothem communities. Recall that just 27 % of the rock wall OTUs was unique, a value indicating significant overlap with the speleothem communities. These analyses suggest that the rock wall community supports many of the same populations found on the adjacent speleothem surfaces.
The SA speleothem communities were quite distinct from those of the BW room as indicated by the Sorabund pairwise comparison analysis (Fig. 2). Proteobacteria were even more abundant in these two communities than in the BW Type 2 group (56 % and 63 %). Further, these Proteobacteria were predominantly Deltaproteobacteria at levels 2 to 3 times greater than speleothem A, the community with the highest abundance of Deltaproteobacteria in the BW room (Fig. 3c). These samples were collected 1 year prior to the BW room samples, thus the unique taxonomic profile observed in this room could reflect both temporal as well as spatial differences, but in either case, the magnitude of variability observed in the community profiles confirms that a range of distinct community types colonize the calcite speleothems in Kartchner Caverns.
Finally, the soil taxonomic composition was distinct from any of the ten cave communities analyzed. While Actinobacteria abundance was similar to that found in the C/D/E/W group, the soil populations were primarily associated with the Rubrobacterales order rather than the Actinomycetales that characterized the majority of the cave Actinobacteria (Fig. 3b). In addition, the abundance of Proteobacteria in the soil sample (7.6 %) was the lowest of all the samples analyzed.
16S rRNA Gene Library of Speleothem D
A 16S rRNA Sanger-sequenced gene library of the speleothem D (BW room, C/D/E/W cluster) bacterial community was constructed to obtain greater phylogenetic resolution of bacterial populations identified in the 454-pyrotag analysis. This speleothem was selected since it belonged to the largest cluster in the BW room and would provide deeper insight into ubiquitous speleothem populations. After quality assessment, 312 clones were obtained that clustered into 57 unique OTUs (97 % similarity cutoff). When compared to the speleothem D pyrotag library, 96 % of the Sanger OTUs were found to have ≥97 % sequence identity with corresponding 454-pyrotags indicating good correspondence between the two methods. Clone 85C, a Gammaproteobacteria that clustered in a lineage with a clone extracted from a grass prairie soil (Fig. 4a, bootstrap, 99), had 100 and 97 % identity to the first and fifth most abundant speleothem D pyrotags, respectively. Clones 43F and 11D had 100 % identity with the second and fourth most abundant pyrotags, respectively. Both of these clones were located in a novel Rubrobacterideae lineage most closely related to the Solirubrobacterales and Thermoleophilales orders (bootstrap, 99). Finally, clone 28G had 100 % identity with the most abundant Kartchner pyrotag, from all samples. This last clone was identified as a member of the Nocardioidaceae family and was located in a strongly supported clade (bootstrap, 100) with Aeromicrobium erythreum, a bacterium known for producing erythromycin [33].


Phylogenetic trees of 16S rRNA gene clone library from Speleothem D. a Phylogenetic tree of gene sequences associated with Proteobacteria classes. b Phylogenetic tree of gene sequences associated with the non-Proteobacteria phyla. The trees topology was determined by maximum parsimony using PAUP version 4.0 Beta 10. The different phylogenetic groups (phyla or class) are delineated by the white and gray areas. Bootstrap values (1,000 replicates) from maximum parsimony analysis are given for nodes with bootstrap values ≥50 %
BLAST analysis revealed that 86 % of the phylotypes identified in the Sanger library were associated with uncultured environmental clones from soil and other cave environments (Supplementary Table 2). Three OTUs clustered in clades (bootstrap 100 %) with clones from other cave systems; clones 2H and 66C, putative Actinobacteria, clustered with a clone from Owl Cave, Kentucky (Q500878) and clone 35E, a putative Deltaproteobacteria, clustered with a clone from Pajsarjeva jama cave, Slovenia (FJ535064) (Fig. 4a and b). In addition, clones 99D, 17G, 21G, 83E, and 98C were found to have ≥99 % sequence identity to Alphaproteobacteria previously cultured from Kartchner Caverns (Fig. 4a and b), four of which were isolated from within the BW room.
The novelty anticipated among cave bacterial populations was confirmed by the fact that 21 % of the clone library OTUs had <96 % similarity to GenBank sequences (Supplementary Table 2). These unique OTUs belonged to a diverse group of phyla including Actinobacteria (11D, 97C and 30D; n = 13), Planctomycetes (29G, 24I, and 31G; n = 6), Alphaproteobacteria (11C; n = 4), Acidobacteria (20C and 4F; n = 2), Chloroflexi (41 F; n = 1), and Gemmatimonadetes (69C; n = 1). Phylogenetic associations observed in this study will guide future efforts to obtain more specific information concerning the potential functional roles of these unknown microbes in the speleothem communities.
Discussion
Diversity and Taxonomic Profile of Speleothem Surfaces
The results from the 454-pyrotag analysis reveal that despite the oligotrophic conditions found in Kartchner Caverns, phylogenetically diverse communities colonize both speleothem and rock surfaces. In fact, two distinct patterns were found among the BW room speleothems. First, a range in both species richness (1,568 to 2,422 OTUs) and diversity (Shannon index, 4.32 to 6.06) was observed, with the C/D/E/W cluster being least diverse and the A and B cluster demonstrating the greatest diversity. The second pattern was the emergence of three distinct phylogenetic clusters from the community taxonomic analysis. Speleothems C/D/E/W were dominated by Actinobacteria, speleothem F was dominated by Acidobacteria and A/B were dominated by Proteobacteria. Taken together, these two patterns reveal that the more diverse communities (A/B) were dominated by Proteobacteria, while the less diverse communities were dominated by Actinobacteria. Overall, these results demonstrate that the variability in speleothem community structure demonstrated previously by our PCR-DGGE study [8] is supported by deep phylogenetic differences among these communities.
The Acidobacteria-dominated community of speleothem F represents an anomaly among the Kartchner communities. Although its richness was intermediate to the C/D/E/W and A/B clusters, and the diversity was not significantly different from C/D/E/W, this community has a unique phylogenetic profile. Previous work has suggested that the physical proximity and the drip water lines feeding each speleothem are possible factors influencing bacterial community structure in Kartchner Caverns [8], but these factors alone do not explain the uniqueness of speleothem F. Recall that this speleothem is located just 0.8 m from D and W. In fact, in contrast to the present pyrotag results, the former PCR-DGGE analysis clustered the F community with D and W [8]. However, PCR-DGGE profiles only document presence/absence of specific populations and do not detect phylogenetic relationships or abundance. Thus, the 454-pyrotag analysis has revealed differences in community composition not evident from the PCR-DGGE analysis. Based on these combined analyses, it appears that while drip water may have an important influence on community structure, there are additional factors shaping the phylogenetic distribution characteristic of each speleothem.
What other factors may be responsible for the unique phylogenetic profile of speleothem F? An elemental analysis of mineral samples removed from the speleothems in 2008, revealed that the surface 2 mm of speleothem F contains between 2.3 and 173 times more Al, Fe, P, Mn, K, Si, Ti, Cr, and Pb than any other formation in the BW room [8]. The major differences observed were in P and Mn with concentrations 7- to 173- and 3.3- to 133 times higher, respectively, for speleothem F than for any other speleothem [8]. These results show that speleothem F was not only unique in its phylogenetic profile but was also unique in its elemental composition. The elemental profile of the speleothems was not identified as an explanatory variable driving variations in overall community structure for the PCR-DGGE analysis, but the elevated levels of these elements in F may explain the unique composition of this particular community. Future work will focus on identifying potential relationships between the unique bacterial profile of speleothem F and factors influencing the metabolism of these elements.
The phylogenetic community types identified in Kartchner Caverns are not unique to this cave, but have been previously identified in limestone cave microbial communities sampled from speleothems, rock walls and soils [11, 34, 35]. Barton et al. [12] found Actinobacteria to be the dominant phylum, representing 60 % of the bacterial community of an oligotrophic limestone rock surface, in Carlsbad Caverns, New Mexico. In other limestone cave studies, Proteobacteria was identified as the dominant phylum, with Alpha-, Gamma-, and Betaproteobacteria classes being most common [11, 36, 37]. Finally, documented abundances of Acidobacteria range from 10 to 33 % in samples taken from cave walls [11], paleolithic paintings [38], cave soil [35], and sediment [34]. Members of these dominant phyla share the characteristics of being well adapted to nutrient-limited growth conditions. Actinobacteria, for example, are known for their ability to grow in nutrient-poor media, their versatility in degrading diverse and recalcitrant humic materials, and their ability to solubilize phosphate and calcium carbonate [7, 39]. In addition, three of the clones identified in the Sanger library were phylogenetically related to clones previously isolated from other cave ecosystems. Novel to this work is the finding that the variability in taxonomic composition found within the BW room on adjacent speleothems exposed to the same microclimate is comparable to the variability observed when comparing geographically diverse caves. This phenomenon suggests that the development of speleothem microbial communities is very sensitive to nutritional inputs and/or to subtle environmental factors and that unique population assemblages can be found on seemingly comparable cave formations. Nevertheless, several dominant OTUs were found in all cave communities evaluated (Supplementary Table 3) thus suggesting that a common core of bacterial communities inhabit cave formations as shown previously [40].
Metabolic Indicator Populations Identified by Sanger and 454 Libraries
Several clones from the speleothem D 16S rDNA gene library identified closely with bacteria having previously documented carbon- and nitrogen-fixing capabilities. First, clone 20H had 99 % sequence identity to Pelomonas saccharophila (Fig. 4), a member of the Betaproteobacteria that has been described as a chemolithoautotrophic hydrogen-oxidizing [41] and nitrogen-fixing bacterium [42]. Second, clone 32E was 99 % similar to Bradyrhizobium japonicum, a nitrogen-fixing bacteria, also described as a versatile facultative chemoautotroph that can grow using H2 as an electron donor [43]. Two other clones (99D and 21G) were included in this well-supported clade (bootstrap, 92) (Fig. 4a). It would be of great interest to determine whether these traditional symbionts can fix nitrogen autotrophically under select environmental conditions as suggested by Franck et al. [44]. A third group of potential autotrophs identified by the 454-pyrotag analysis, though not detected in the Sanger library, were those belonging to the Nitrospirales order, representing 0.1 to 5 % of the OTUs in every speleothem community. This order includes an abundance of obligate chemolithotrophic phylotypes belonging to the genera Nitrospira and Leptospirillum, bacteria that obtain energy from nitrite [45] and iron [46] oxidation. Although the specific metabolism of these OTUs could not be confirmed from phylogenetic associations, the activity of both of these genera is acid generating, a process that has been shown to enhance the solubilization of nutrients from primary minerals [47]. The most abundant member of the Nitrospira in the CA and ST communities was 98 % similar to a Nitrospirales isolate from a cave in Slovenia (FJ535150).
The majority of the Sanger OTUs analyzed were associated with heterotrophic bacteria in the phylogenetic analysis (Fig. 4a and b). Heterotrophic OTUs of particular interest were those previously associated with oligotrophic conditions. Betaproteobacteria clone 22B, the third most abundant phylotype in the Sanger library (n = 23 sequences), was 98 % similar to Polaromonas aquatica. Polaromonas species are commonly found in cold and nutrient-poor environments such as glacial melt waters [48] and oligotrophic water systems [49]. Alphaproteobacteria clone 98C was 99 % similar to Sphingopyxis alaskensis (CP000356), a classic marine heterotrophic ultramicrobacterium used to model oligotrophy [50, 51]. This clone was also 99.8 % similar to a bacterium previously isolated from a different region of Kartchner Caverns (DQ205308) [52]. Though metabolic potential cannot be confirmed from phylogenetic associations, this analysis provides critical information to guide future efforts to culture these organisms.
A final taxonomic group of interest were the third and eighth most abundant pyrotags in the 454-library (Supplementary Table 3) that were identified as close relatives to the genus Sporichthya, a rare heterotrophic Actinobacteria whose growth is stimulated when a combination of gellan gum (a solidifying agent) and CaCl2 are added to the media [53]. The implication that the metabolism of this highly abundant OTU could be stimulated by Ca in this limestone cave is an intriguing possibility especially in light of several studies that have demonstrated the ability of cultured cave bacteria to precipitate calcium carbonate in vitro [54–56].
In conclusion, the robust diversity analysis conducted in the BW room of Kartchner Caverns provides key information that will be needed for future efforts to characterize the energy dynamics driving the formation of these subterranean communities. In fact, results from the 454-pyrotag and the Sanger clone library have provided clues about potential energy sources in the cave including nitrite and hydrogen oxidation. Further, the Sanger clone library revealed the presence of clones closely associated with bacteria with carbon- and nitrogen-fixing capabilities as well as specific adaptations to oligotrophy. This information serves as a starting point for current functional metagenomic efforts in Kartchner Caverns, targeting adjacent speleothems, to determine the energy drivers of this ecosystem as well as to elucidate links between taxonomic and functional diversity of cave formations.
References
Sarbu SM, Kinkle BK, Vlasceanu L, Kane TC, Popa R (1994) Microbiological characterization of a sulfide-rich groundwater ecosystem. Geomicrobiol J 12:175–182
Jones DS, Tobler DJ, Schaperdoth I, Mainiero M, Macalady JL (2010) Community structure of subsurface biofilms in the thermal sulfidic caves of Acquasanta Terme, Italy. Appl Environ Microbiol 76:5902–5910
Cunningham KI, Northup DE, Pollastro RM, Wright WG, Larock EJ (1995) Bacteria, fungi and biokarst in Lechuguilla Cave, Carlsbad Caverns National Park, New Mexico. Environ Geol 25:2–8
Northup DE, Barns SM, Yu LE, Spilde MN, Schelble RT, Dano KE, Crossey LJ, Connolly CA, Boston PJ, Natvig DO, Dahm CN (2003) Diverse microbial communities inhabiting ferromanganese deposits in Lechuguilla and Spider Caves. Environ Microbiol 5:1071–1086
Hill CA, Forti P (1997) Kartchner Caverns, Arizona. In: Hill CA, Forti P (eds) Cave minerals of the world. National Speleological Society, Huntsville, AL, pp 350–354
Levy DB (2007) Oxidation-reduction chemistry of Lechuguilla Cave seepage. J Cave Karst Stud 69:351–358
Laiz L, Groth I, Gonzalez I, Saiz-Jimenez C (1999) Microbiological study of the dripping waters in Altamira cave (Santillana del Mar, Spain). J Microbiol Meth 36:129–138
Legatzki A, Ortiz M, Neilson JW, Casavant RR, Palmer MW, Rasmussen C, Pryor BM, Pierson LS, Maier RM (2012) Factors influencing observed variations in the structure of bacterial communities on calcite formations in Kartchner Caverns, AZ, USA Geomicrobiol J 29:422–434
Legatzki A, Ortiz M, Neilson JW, Dominguez S, Andersen GL, Toomey RS, Pryor BM, Pierson LS, Maier RM (2011) Bacterial and archaeal community structure of two adjacent calcite speleothems in Kartchner Caverns, Arizona, USA. Geomicrobiol J 28:99–117
Laiz L, Groth I, Schumann P, Zezza F, Felske A, Hermosin B, Saiz-Jimenez C (2000) Microbiology of the stalactites from Grotta dei Cervi, Porto Badisco, Italy. Int Microbiol 3:25–30
Schabereiter-Gurtner C, Saiz-Jimenez C, Pinar G, Lubitz W, Rolleke S (2004) Phylogenetic diversity of bacteria associated with Paleolithic paintings and surrounding rock walls in two Spanish caves (Llonin and La Garma). FEMS Microbiol Ecol 47:235–247
Barton HA, Taylor NM, Kreate MP, Springer AC, Oehrle SA, Bertog JL (2007) The impact of host rock geochemistry on bacterial community structure in oligotrophic cave environments. Int J Speleol 36:93–104
Buecher RH (1999) Microclimate study of Kartchner Caverns, Arizona. J Cave Karst Stud 61:108–120
Buecher DC, Sidner RM (1999) Bats of Kartchner Caverns State Park. Arizona J Cave Karst Stud 61:6
Welbourn WC (1999) Invertebrate cave fauna of Kartchner Caverns, Arizona. J Cave Karst Stud 61:93–101
Drees KP, Neilson JW, Betancourt JL, Quade J, Henderson DA, Pryor BM, Maier RM (2006) Bacterial community structure in the hyperarid core of the Atacama Desert, Chile. Appl Environ Microbiol 72:7902–7908
Solis-Dominguez FA, Valentin-Vargas A, Chorover J, Maier RM (2011) Effect of arbuscular mycorrhizal fungi on plant biomass and the rhizosphere microbial community structure of mesquite grown in acidic lead/zinc mine tailings. Sci Total Environ 409:1009–1016
Baker GC, Smith JJ, Cowan DA (2003) Review and re-analysis of domain-specific 16S primers. J Microbiol Meth 55:541–555
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Sun YJ, Cai YP, Liu L, Yu FH, Farrell ML, McKendree W, Farmerie W (2009) ESPRIT: estimating species richness using large collections of 16S rRNA pyrosequences. Nucleic Acids Res 37:e76
Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere”. P Natl Acad Sci USA 103:12115–12120
Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML (2008) Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet 4:e1000255
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Lane D (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, Chichester, UK, pp 115–173
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar BA, Lai T, Steppi S, Jobb G, Forster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, Konig A, Liss T, Lussmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer KH (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371
Swofford DL (2006) PAUP* 4.0 Phylogenetic analysis using parsimony, Macintosh Beta v.10. Sinauer, Sunderland
Bond PL, Hugenholtz P, Keller J, Blackall LL (1995) Bacterial community structures of phosphate-removing and non-phosphate-removing activated sludges from sequencing batch reactors. Appl Environ Microbiol 61:1910–1916
Militon C, Boucher D, Vachelard C, Perchet G, Barra V, Troquet J, Peyretaillade E, Peyret P (2010) Bacterial community changes during bioremediation of aliphatic hydrocarbon-contaminated soil. FEMS Microbiol Ecol 74:669–681
Papineau D, Walker JJ, Mojzsis SJ, Pace NR (2005) Composition and structure of microbial communities from stromatolites of Hamelin Pool in Shark Bay, Western Australia. Appl Environ Microbiol 71:4822–4832
Miller ES, Woese CR, Brenner S (1991) Description of the erythromycin-producing bacterium Arthrobacter sp. Strain Nrrl-B-3381 as Aeromicrobium erythreum gen. nov., sp. nov. Int J Syst Bacteriol 41:363–368
Chelius MK, Moore JC (2004) Molecular phylogenetic analysis of Archaea and Bacteria in Wind Cave, South Dakota. Geomicrobiol J 21:123–134
Zhou JP, Gu YQ, Zou CS, Mo MH (2007) Phylogenetic diversity of bacteria in an earth-cave in Guizhou Province, Southwest of China. J Microbiol 45:105–112
Barton HA, Taylor RM, Pace NR (2004) Molecular phylogenetic analysis of a bacterial community in an oligotrophic cave environment. Geomicrobiol J 21:11–20
Schabereiter-Gurtner C, Saiz-Jimenez C, Pinar G, Lubitz W, Rolleke S (2002) Altamira cave Paleolithic paintings harbor partly unknown bacterial communities. FEMS Microbiol Lett 211:7–11
Zimmermann J, Gonzalez JM, Saiz-Jimenez C, Ludwig W (2005) Detection and phylogenetic relationships of highly diverse uncultured acidobacterial communities in Altamira cave using 23S rRNA sequence analyses. Geomicrobiol J 22:379–388
El-Tarabily KA, Nassar AH, Sivasithamparam K (2008) Promotion of growth of bean (Phaseolus vulgaris L.) in a calcareous soil by a phosphate-solubilizing, rhizosphere-competent isolate of Micromonospora endolithica. Appl Soil Ecol 39:161–171
Porca E, Jurado V, Zgur-Bertok D, Saiz-Jimenez C, Pasic L (2012) Comparative analysis of yellow microbial communities growing on the walls of geographically distinct caves indicates a common core of microorganisms involved in their formation. FEMS Microbiol Ecol 81:255–266
Doudoroff M (1940) The oxidative assimilation of sugars and related substances by Pseudomonas saccharophila with a contribution to the problem of the direct respiration of di- and polysaccharides. Enzymologia 9:59–72
Barraquio WL, Padre BC, Watanabe I, Knowles R (1986) Nitrogen fixation by Pseudomonas saccharophila Doudoroff ATCC 15946. J Gen Microbiol 132:237–241
Hanus FJ, Maier RJ, Evans HJ (1979) Autotrophic growth of H2-uptake-positive strains of Rhizobium japonicum in an atmosphere supplied with hydrogen gas. P Natl Acad Sci USA 76:1788–1792
Franck WL, Chang WS, Qiu J, Sugawara M, Sadowsky MJ, Smith SA, Stacey G (2008) Whole-genome transcriptional profiling of Bradyrhizobium japonicum during chemoautotrophic growth. J Bacteriol 190:6697–6705
Ehrich S, Behrens D, Lebedeva E, Ludwig W, Bock E (1995) A new obligately chemolithoautotrophic, nitrite-oxidizing bacterium. Nitrospira moscoviensis sp. nov. and its phylogenetic relationship. Arch Microbiol 164:16–23
Hippe H (2000) Leptospirillum gen. nov. (ex Markosyan 1972), nom. rev., including Leptospirillum ferrooxidans sp. nov. (ex Markosyan 1972), nom. rev. and Leptospirillum thermoferrooxidans sp. nov. (Golovacheva et al. 1992). Int J Syst Evol Microbiol 50:501–503
Sonnleitner R, Redl B, Merschak P, Schinner F (2011) Mobilization of metals from pristine mineral soil by nitrifying and sulfur-oxidizing bacteria—the leaching potential of indigenous culture enrichments. Geomicrobiol J 28:212–220
Irgens RL, Gosink JJ, Staley JT (1996) Polaromonas vacuolata gen. nov., sp. nov., a psychrophilic, marine, gas vacuolate bacterium from Antarctica. Int J Syst Bacteriol 46:822–826
Loy A, Beisker W, Meier H (2005) Diversity of bacteria growing in natural mineral water after bottling. Appl Environ Microbiol 71:3624–3632
Lauro FM, McDougald D, Thomas T, Williams TJ, Egan S, Rice S, DeMaere MZ, Ting L, Ertan H, Johnson J, Ferriera S, Lapidus A, Anderson I, Kyrpides N, Munk AC, Detter C, Han CS, Brown MV, Robb FT, Kjelleberg S, Cavicchioli R (2009) The genomic basis of trophic strategy in marine bacteria. Proc Natl Acad Sci USA 106:15527–15533
Ostrowski M, Fegatella F, Wasinger V, Guilhaus M, Corthals GL, Cavicchioli R (2004) Cross-species identification of proteins from proteome profiles of the marine oligotrophic ultramicrobacterium, Sphingopyxis alaskensis. Proteomics 4:1779–1788
Ikner LA, Toomey RS, Nolan G, Neilson JW, Pryor BM, Maier RM (2007) Culturable microbial diversity and the impact of tourism in Kartchner Caverns, Arizona. Microb Ecol 53:30–42
Suzuki S, Okuda T, Komatsubara S (1999) Selective isolation and distribution of Sporichthya strains in soil. Appl Environ Microbiol 65:1930–1935
Cacchio P, Contento R, Ercole C, Cappuccio G, Martinez MP, Lepidi A (2004) Involvement of microorganisms in the formation of carbonate speleothems in the Cervo Cave (L’Aquila-Italy). Geomicrobiol J 21:497–509
Sanchez-Moral S, Canaveras JC, Laiz L, Saiz-Jimenez C, Bedoya J, Luque L (2003) Biomediated precipitation of calcium carbonate metastable phases in hypogean environments: a short review. Geomicrobiol J 20:491–500
Danielli HMC, Edington MA (1983) Bacterial calcification in limestone caves. Geomicrobiol J 3:1–16
Acknowledgments
We would like to express our appreciation to Robert Casavant and Ginger Nolan for their assistance during speleothem selection and sampling in Kartchner Caverns. Also, we want to thank Nick Sisneros for his assistance with 454 pyrosequencing. Funding for this work was supplied by the National Science Foundation Microbial Observatory grant MCB0604300.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 276 kb)
Rights and permissions
About this article
Cite this article
Ortiz, M., Neilson, J.W., Nelson, W.M. et al. Profiling Bacterial Diversity and Taxonomic Composition on Speleothem Surfaces in Kartchner Caverns, AZ. Microb Ecol 65, 371–383 (2013). https://doi.org/10.1007/s00248-012-0143-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-012-0143-6