
Abstract
Members of the rare microbiome can be important components of complex microbial communities. For example, pet dog ownership is a known risk factor for human campylobacteriosis, and Campylobacter is commonly detected in dog feces by targeted assays. However, these organisms have not been detected by metagenomic methods. The goal of this study was to characterize fecal microbiota from healthy and diarrheic pet dogs using two different levels of molecular detection. PCR amplification and pyrosequencing of the universal cpn60 gene target was used to obtain microbial profiles from each dog. To investigate the relatively rare epsilon-proteobacteria component of the microbiome, a molecular enrichment was carried out using a PCR that first amplified the cpn10–cpn60 region from epsilon-proteobacteria, followed by universal cpn60 target amplification and pyrosequencing. From the non-enriched survey, the major finding was a significantly higher proportion of Bacteroidetes, notably Bacteroides vulgatus, in healthy dogs compared to diarrheic dogs. Epsilon-proteobacteria from the genera Helicobacter and Campylobacter were also detected at a low level in the non-enriched profiles of some dogs. Molecular enrichment increased the proportion of epsilon-proteobacteria sequences detected from each dog, as well as identified novel, presumably rare sequences not seen in the non-enriched profiles. Enriched profiles contained known species of Arcobacter, Campylobacter, Flexispira, and Helicobacter and identified two possibly novel species. These findings add to our understanding of the canine fecal microbiome in general, the epsilon-proteobacteria component specifically, and present a novel modification to traditional metagenomic approaches for study of the rare microbiome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most natural microbial environments are complex, with a distribution of organisms that can span several orders of magnitude in abundance. The term “rare microbiome” is often applied to the organisms that are detected infrequently or at levels close to the detection limit of the methodology used. Microbial ecology studies have greatly benefited from the advent of pyrosequencing, which allow for the direct sequencing of millions of PCR products in a single run. Despite this increase in sequencing depth, datasets can contain operational taxonomic units (OTUs) derived from a single sequencing read. There is debate as to whether these rare OTUs truly represent rare organisms in the community or if a significant number are derived from technical errors [1, 2].
One environment that has been studied at different levels of resolution is the dog fecal microbiome. Dogs are part of the North American family, with a pet dog found in one third of American and Canadian households [3, 4]. Campylobacteriosis, a diarrheal illness caused by Campylobacter spp., has been repeatedly linked to pet dog ownership [5–7]. Culture-based studies of Campylobacter shedding in dog feces have reported that 2.7–100% of dogs shed multiple Campylobacter species [8–12]. Using targeted quantitative PCR (qPCR) assays, the same picture emerges, with 50–100% of dogs shedding detectable levels of multiple Campylobacter species [13–15]. Diarrhea has been associated with increased Campylobacter shedding and increased species richness of the Campylobacter spp. detected [15]. Collectively, this body of work firmly establishes Campylobacter as a common member of the dog large intestinal/fecal microbiota.
The composition of canine large intestine/fecal microbiota has also been investigated as a collective population. 16S rRNA sequence profiles have been generated and compared to the existing picture of the dog fecal microbiota based on culture, fluorescent in situ hybridization (FISH), terminal restriction fragment length polymorphism (T-RFLP), and denaturing gel gradient electrophoresis (DGGE) [13, 14, 16]. Unamplified metagenomic sequencing has also recently been performed [17]. Sequence profiles indicate that Firmicutes and Bacteroidetes (and occasionally Fusobacteria) are the dominant phyla, with Proteobacteria making up a minor component (<15%) [17–19]. These results generally agree with culture and FISH estimates [20–24]. Campylobacter (epsilon-proteobacteria) has not been reported from these studies, which is not surprising given that Campylobacter spp. are estimated to be in the range of 103–108 organisms/g of feces while total fecal bacteria can exceed 1010 organisms/g [15, 20]. As such, although Campylobacter spp. are expected to be part of the microbiome, they usually occur in quantities below the detection limit of the population-level methods used.
The goal of our study was to use pyrosequencing to investigate the epsilon-proteobacteria populations in healthy and diarrheic domestic pet dogs in the context of the total fecal microbiome. Non-enriched microbial profiles for healthy and diarrheic dogs were determined by PCR and pyrosequencing of the cpn60 gene, which has been shown to have superior resolution compared to the 16S rRNA gene [25]. In parallel, each sample was also subjected to a molecular enrichment for epsilon-proteobacteria sequences by class-specific PCR of the cpn10–cpn60 gene region followed by universal cpn60 PCR. We observed dramatic differences in the Bacteroidetes community of healthy and diarrheic dogs (regardless of the etiology of the diarrhea), while enrichment profiles exposed an order of magnitude more diversity in epsilon-proteobacteria sequences compared to their non-enriched profile counterparts.
Materials and Methods
Sample Collection and DNA Extraction
Fecal samples from healthy pet dogs and dogs with diarrhea (of any etiology) were collected as part of a previous study [15] (Table 1). Samples were stored at −80°C until total bacterial DNA was extracted using the QIAamp DNA stool kit (Qiagen), following all the recommended steps for “Isolation of DNA from stool for pathogen detection.” Final DNA samples were diluted 1:10 with sterile water before PCR.
cpn60 Universal Target PCR
The universal target (UT) region of the cpn60 gene was amplified using a primer cocktail as described previously [25–27]. Primer sets were modified at the primers 5′ end with one of ten unique 10-mer multiplexing identification (MID) sequences as per the manufacturer’s recommendations (454 Life Sciences, Branford, CT, USA). PCR consisted of 1× PCR reaction buffer (20 mM Tris–HCl (pH 8.4), 50 mM KCl), 2.5 mM MgCl2, 200 μM dNTP, 400 nM of forward and reverse primer, 2.5 U Platinum Taq DNA polymerase (Invitrogen, Burlington, ON, Canada) and 2 μl template DNA, in a final volume of 50 μl. Eight to 12 PCR reactions were run for each sample in a thermocycler (Eppendorf Mastercycler or BioRad MyiQ) over a temperature gradient: 94°C for 3 min, followed by 40 cycles of 30 s at 94°C, 1 min at 42–60°C, and 1 min at 72°C, followed by a final extension at 72°C for 10 min. PCR reactions from the same sample were pooled and concentrated using the AMPure Purification system (Agencourt Bioscience, Beverly, MA, USA) and purified by agarose gel separation and extraction (QIAEX II gel extraction kit, Qiagen). Final amplicon was suspended in TE buffer (10 mM Tris–HCl, 1 mM EDTA; pH 8.0) and quantified using a Qubit fluorometer (Invitrogen).
Epsilon-Proteobacteria Enrichment
The cpn10–cpn60 operon (coding region plus inter-spacer region) from 49 proteobacteria, comprised of six alpha-, six beta-, six gamma-, 12 delta- and 19 epsilon-proteobacteria (Supplemental Table 1), were aligned using ClustalW [28] (gap opening penalty of 50 and gap extension penalty of five). Consensus regions that included epsilon-proteobacteria sequences and excluded all others were identified (Supplemental Figure 1) and primers JH0108 (forward; 5′-ATG AAN TTT CAR CCW YTW GG-3′), and JH0102 (reverse; 5′-ARC ATH KCT TTT CTT CTR TC-3′), corresponding to nucleotides 1–20 of the E. coli cpn10 sequence and nucleotides 847–866 of the E. coli cpn60 sequence, were designed.
Genomic DNA from Campylobacter jejuni subsp. jejuni, Campylobacter fetus, Helicobacter nemestrinae, Salmonella sp., Pseudomonas aeruginosa, Bartonella sp., and Pasteurella haemolytica was used to optimize the epsilon-enrichment primer conditions. Magnesium gradients (data not shown) and temperature gradients (Supplemental Figure 2) were tested to determine that the specific cpn10–cpn60 amplicon (∼1,200 bp) could be generated from the epsilon-proteobacteria species (Campylobacter and Helicobacter samples) to the exclusion of the alpha-proteobacteria (Bartonella) and gamma-proteobacteria (Salmonella, Pseudomonas, and Pasteurella) species tested. Only epsilon-proteobacteria species generated a band at ∼1,200 bp (Supplemental Figure 2).
The epsilon-proteobacteria enrichment of dog fecal DNA extracts was carried out by PCR amplification using 1× PCR reaction buffer [20 mM Tris–HCl (pH 8.4), 50 mM KCl], 5 mM MgCl2, 200 μM dNTP, 400 nM of JH0108 and JH0102, 2.5 U Platinum Taq DNA Polymerase (Invitrogen), and 2 μl template DNA, in a final volume of 50 μl, using the program: 94°C for 3 min, followed by 40 cycles of 30 s at 94°C, 1 min at 50°C, and 2 min at 72°C, followed by a final extension at 72°C for 10 min. The entire PCR reaction was visualized on an agarose gel with ethidium bromide staining and UV exposure and the band at ∼1,200 bp was extracted and purified (QIAEX II gel extraction kit, Qiagen). Gel-purified DNA was subsequently used as template DNA in cpn60 UT PCR as described above.
454 Pyrosequencing
For high throughput sequencing using the GS FLX Titanium system (Roche), cpn60 UT amplicon libraries were pooled in equimolar concentrations as groups of five and run within eight regions of a 16-region gasketed plate. Emulsion PCR and sequencing was performed at the National Research Council Plant Biotechnology Institute, Saskatoon, SK.
Quality Control and Assembly of OTU
Pyrosequencing data was processed using the default on-rig procedures from 454/Roche (Branford, CT, USA), and filter-passing reads were partitioned by MID. For most reads, a recognizable MID was identified and the read partitioned using the SFFtools from Roche/454. Additional reads that exhibited partial degradation of the MID were recovered using custom PERL scripts. The recovery of reads with partially degraded MIDs was performed by identifying the cpn60 UT primer site within each read and capturing the sequence immediately 5′ of the cpn60 UT primer. This subsequence (<10 bp) is a perfect suffix of the MID sequence and can be traced to its appropriate MID with surety given the set of MIDs loaded in the same physical region. OTUs were assembled from the entire dataset (37 profiles total) using the complementary DNA mode for Newbler (version 2.3), an overlap MinMatchIdentity of 91%, and overlap MinMatchLength of 137 (which corresponds to at least 25% of the cpn60 UT), as previously optimized for cpn60 amplicons [25, 27]. During assembly, “outlier” sequences (including possible chimeras, 4,724 reads) and reads of insufficient length for assembly were removed from further analysis. Nearest neighbor taxonomic labels were assigned to each OTU based on the best match to reference sequences from cpnDB [29] as determined by watered-BLAST [25]. Post-assembly quality control steps included removal of OTUs having <60% identity to any cpn60 sequence in the reference database (manually confirmed to represent non-specific amplification products). OTU sequences are available from cpnDB (http://www.cpndb.ca), and detailed information about each OTU is listed in Supplementary Table 2.
Data Analysis
Rarefraction analysis and diversity statistics (Shannon diversity index, Simpson diversity index, bias-corrected Chao1 richness estimate, and Good’s coverage estimate) for each library were calculated from actual read counts for each OTU using mothur [30]. When sample profiles were compared directly to each other, read counts for each OTU were scaled to a median library size of 4,491 reads. Evaluation of the relatedness between sample profiles was performed using Unifrac [31, 32]. The input phylogenetic tree required for Unifrac analysis was created from full-length cpn60 UT reference sequences representing the nearest neighbor of each OTU. All phylogenetic trees were constructed from ClustalW sequence alignments [28] with a gap opening penalty of 50 and gap extension penalty of 5 before trees were constructed using PHYLIP [33]. A DNA distance matrix was calculated based on the F84 maximum likelihood option and a neighbor-joined tree was assembled. Weighted Unifrac distances were calculated, and jackknife clustering was performed for all sample profiles together and non-enriched profiles alone.
Statistical Analysis
To determine if there were significant differences in diversity statistics between healthy and diarrheic samples or non-enriched and enriched samples, as well as between phyla or species detected, Levene’s test was used to determine equality of variances, followed by either an independent t test (for equal variances) or a Mann–Whitney U test (for unequal variances) using SPSS software (SPSS Inc., Chicago, IL, USA). A p < 0.05 was used as a cutoff for statistical significance.
Results
Sample Profile Generation
Thirty-seven cpn60 amplicon libraries were prepared and sequenced from 18 fecal samples [nine from healthy dog samples (HDS), nine from diarrheic dog samples (DDS)]. Non-enriched cpn60 profiles and epsilon-enriched profiles were produced for each sample. One technical replicate of an epsilon-enriched profile was performed (for sample HDS1fall). A total of 229,981 MID-labeled sequencing reads were recovered with an average of 6,216 reads per sample (range, 102–22,411; median, 4,491). Fewer than 1,000 reads were obtained for five samples [HDS9A non-enriched (783 reads) and enriched (793), DDS11 non-enriched (102) and enriched (165) and HDS1spring enriched (599)]. It should be noted that low sequence read counts appeared to be sample-related (mainly from HDS9A and DDS11), which suggests that something about these samples may have affected the efficacy of the pyrosequencing protocol. Downstream analyses of the aggregate data was performed both with and without the low sequence read profiles, and since there were no significant differences in either the trends or statistical results obtained, all of the samples were included in the following analyses.
Canine Fecal Microbiome Characteristics
A total of 834 cpn60 OTUs were assembled from the entire dataset. Non-enriched healthy dog profiles contained 22–183 OTU (median richness of 109 OTU), non-enriched diarrheic dog profiles contained 10–160 OTU (median richness of 74 OTU), epsilon-enriched healthy dog profiles contained 24–144 OTU (median richness of 56 OTU), and epsilon-enriched diarrheic dog profiles contained 17–243 OTU (median richness of 36 OTU). There were no statistically significant differences (at the alpha = 5% level of significance) in observed number of OTU between sample type groups, but the trend was for the non-enriched profiles to have more OTU than the epsilon-enriched profiles and for healthy dogs to have more OTU than diarrheic ones (Table 2). This trend continued when the bias-corrected Chao1 richness estimate was calculated: non-enriched healthy dog profiles had the greatest OTU richness, while the epsilon-enriched diarrheic profiles had the least (Table 2). Good’s coverage estimates ranged from 0.916 (HDS1spring enriched profile) to 0.999 (DDS64 non-enriched profile) (Supplementary Figure 3), with an average sample coverage of 0.986 ± 0.018.
The same trends were observed with diversity indicators as were seen with richness indicators. Shannon diversity and Simpson’s reciprocal indices were highest for non-enriched healthy dog profiles, followed by non-enriched diarrheic dog profiles and epsilon-enriched healthy dog profiles, with epsilon-enriched diarrheic dog profiles being the least diverse (Table 2).
Clustering of profiles by Unifrac distance revealed that both health status and profile type (non-enriched or enriched) were the major influences on the similarity between sample profiles. Jackknifed clustering of non-enriched cpn60 profiles distinguished clusters primarily by health status (healthy or diarrheic) (Supplemental Figure 4A). However, this distinction was not completely clear, with DDS18 clustering with healthy samples and HDS19 and HDS9A clustering with diarrheic samples. When all profiles were analyzed together, two major clusters were formed with non-enriched profiles in one group and epsilon-enriched profiles in the other. Two of the low sequence read profiles (HDS9A non-enriched and DDS11 enriched) were outliers (Supplemental Figure 4B).
Non-Enriched Profiles of Healthy and Diarrheic Dogs
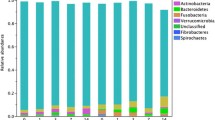
Non-enriched profiles from all 18 samples were examined individually (Fig. 1a) and as a collective by health status (Fig. 1b). Healthy dogs had a dominant Bacteroidetes population (∼50% of reads), with the remainder of the community composed of Firmicutes and Proteobacteria (∼25% each). The diarrheic profiles showed a significant shift away from Bacteroidetes, instead having a Firmicutes/Proteobacteria/Bacteroidetes/Actinobacteria ratio of 4:4:1:1 (Fig. 1b). Interestingly, the only phylum that was significantly different in abundance between health states was the Bacteroidetes (p << 0.05). In the diarrheic profiles, Bacteriodetes was not simply replaced by another phylum, but was compensated for by generally higher abundances of Firmicutes or Proteobacteria, depending on the dog. Epsilon-proteobacteria were detected at low levels in the Proteobacteria portion of the non-enriched profiles of healthy dogs (approximately 11% of the Proteobacteria; Fig. 1b). Proteobacteria comprised a larger proportion of the diarrheic dog profiles (average 36.1%), but the epsilon-proteobacteria were almost undetected (<0.3% of the Proteobacteria).

Composition of the universal dog fecal profiles at the phylum level by a individual dog and b combined health status (n = 9)
The majority of the Bacteroidetes OTUs detected in this study belonged to the genus Bacteroides. Since the cpn60 UT region has the discriminating power to speciate Bacteroides [34], an analysis of Bacteroides species was conducted (Table 3). Within the healthy dog population, the most abundant Bacteroides sequences were B. vulgatus, both in terms of the number of OTU and number of normalized sequence reads per dog (Table 3). In the diarrheic dogs, significantly fewer B. vulgatus-like reads and B. vulgatus OTUs were detected (Table 3).
Epsilon-Proteobacteria in Healthy and Diarrheic Dogs
PCR primers flanking the cpn60 UT region were designed to amplify epsilon-proteobacteria and used in a nested-PCR protocol. To determine the reproducibility of this technique, replicate enrichments of sample HDS1fall were performed. The composition of the replicate profiles consisted of Actinobacteria/Bacteroidetes/Firmicutes/Proteobacteria in the proportions of <1:<1:18:82 and <1:<1:6:93 for the two replicates (Supplemental Figure 5). When the replicates were compared at the genus level, the dominant genus (by an order of magnitude) was Helicobacter in both profiles, with only minor differences in rare genera detected (Supplemental Figure 5). The weighted Unifrac distance between HDS1fall technical replicates was 0.107, the smallest distance obtained for any profile comparison in this study. By comparison, the average weighted Unifrac distance between all enriched profiles was 0.475 ± 0.137.
Epsilon-proteobacteria were detected at low levels in the non-enriched libraries of both healthy and diarrheic dogs, with healthy dog samples containing 40 detectable epsilon-proteobacteria OTUs (2.66% of reads) and diarrheic dog samples containing 11 epsilon-proteobacteria OTUs (0.02% of reads) (Table 4). When the same samples were enriched for epsilon-proteobacteria sequences, the healthy and diarrheic dog populations contained 212 and 152 epsilon-proteobacteria OTUs, accounting for 81.81% and 23.19% of the reads, respectively (Table 4). This increase in OTU numbers and percent composition was contributed to by both increased detection of OTUs observed in the non-enriched profiles (11 instances) and the detection of new OTUs (six instances) (Table 4). The latter cases highlight the success of molecular enrichment for detection of rare species. For example, Arcobacter skirrowii was not detected in any dog in any of the non-enriched profiles. After enrichment, two distinct OTU belonging to A. skirrowii were detected in both healthy and diarrheic dog samples, indicating that this species was present below the detection limit of the non-enriched profiles generated (Table 4). There was only a single case (Campylobacter concisus) where an OTU was detected in the non-enriched profile and not in the enriched profile. In addition, a collection of Helicobacter sequences distinct from the reference sequence collection of Helicobacter species was detected in all four sample pools (Table 4). This Helicobacter “species” was distinct from reference sequences by pairwise identity (87.3–94.8%), branched distinctly by phylogenetic analysis (Supplemental Figure 6) and was the most dramatically increased in representation by the enrichment protocol (Table 4).
Discussion
The dog fecal microbiome is a potential indicator of animal health and also acts as a reservoir for zoonotic pathogens. In attempting to better understand the structure of this microbial community, a variety of methods, including culture, FISH, T-RFLP, DGGE, metagenomic sequencing, and 16S rRNA-based pyrosequencing, have been used to characterize the fecal microbiota of both laboratory and pet dogs during health, episodes of diarrhea, with dietary modifications or the administration of antibiotics [13, 14, 17–20, 22–24]. In many cases, when specific groups of organisms, like Campylobacter or Enterococcus, are of interest, studies were supplemented with specific PCR or qPCR to quantify known species [13, 14]. This is often because organisms of special interest are part of the rare microbiome and cannot be detected with the over-arching survey methods used. Of all the methods available, deep sequencing of samples by pyrosequencing offers the advantage of collecting specific sequence information from a broad range of organisms, including the “unculturables.” New sequencing instruments have been introduced that offer more potential for deeper community profiling and greater direct access to parts of the rare microbiome. However, depending on the complexity of the community and its rank abundance profile, even this level of sequencing may not be sufficient to detect very rare constituents, and furthermore, the approach may constitute “overkill” depending on the question being investigated. For researchers with a targeted question, an enrichment approach like the one presented here offers a solution based on resources and tools that are widely available.
The goal of this study was to investigate the fecal microbiota of healthy and diarrheic dogs by pyrosequencing the cpn60 gene target. This target, which encodes the type I chaperonin protein, has been used successfully for the characterization of the cat fecal microbiome [35], as well as other intestinal environments [36–38]. The cpn60 UT has been demonstrated to allow better resolution of closely related taxa compared with the 16S rRNA target [25] while overcoming known limitations of 16S rRNA-based detection (such as under-representation of bifidobacteria) [38]. cpn60 UT PCR protocols for faithful community representation have been validated [26], compared to pyrosequenced community profiles based on 16S rRNA gene targets [25], and are supported by a high-quality reference sequence database [29]. In addition to overall community profiling, we investigated the utility of introducing a molecular enrichment step prior to universal PCR to enrich the resulting profiles for sequences from rare organisms without the need to limit our results to previously characterized species. The epsilon-proteobacteria was targeted for this purpose to gain a better understanding of the Campylobacter and Helicobacter species present and possibly identify novel taxa within this class.
The diversity of the fecal microbiota was not significantly different between healthy and diarrheic dogs in this study (Table 2). There was a non-significant trend for healthy dogs to have more OTU and slightly higher Shannon and Simpson’s diversity indices than their diarrheic counterparts, but these differences were offset by large animal-to-animal variation. Previous study of this collection of samples determined that the total 16S rRNA copies and cpn60 copies detectable per gram of feces was approximately the same between healthy and diarrheic feces, indicating that the diarrheic animals did not shed more or fewer bacteria than healthy ones [15]. In this study, we are able to elucidate that the difference between health and disease states is coincident not in terms of OTU diversity, but rather in the taxonomic composition of the microbial communities (Fig. 1). This phenomenon has been seen before in dogs, in a study in which diet differences were associated with different taxonomic profiles but comparable numbers of OTU [19].
Examining the fecal microbiota profiles in Fig. 1a, one healthy dog (HDS9A) and one diarrheic dog (DDS18) appear noticeably out of place. Healthy dog HDS9A has a profile almost entirely composed of Firmicutes sequences. Interestingly, this was the only dog in this study that was fed a vegetarian diet (Table 1). While this profile was also one of the low sequence read profiles (783 reads), there is sufficient data to reflect the overall composition of the microbiota. Further study is needed to determine if a Firmicutes-dominated microbiota is characteristic of a vegetarian diet or if this profile is simply a dog-specific observation. The other anomalous profile was DDS18, which looked strikingly characteristic of a healthy dog. Upon review of the case, it was noted that dogs DDS18 and DDS19 were littermates residing in the same household (Table 1). Fecal samples for DDS18 and DDS19 were submitted for parasitology testing from both dogs when diarrhea was detected in the household but not resolved to the level of individual dog. Based on the fecal profiles generated in this study, we hypothesize that only DDS19 had diarrhea at the time of sampling and that DDS18 was included as a diarrheic dog sample in error or to test it for subclinical infection. A larger study of healthy and diarrheic dog fecal profiles is necessary to be able to draw broad conclusions, but this small study does reveal trends that create testable hypotheses for further investigations.
A common question raised during microbial ecology studies is how stable a population is over time. While the majority of the original healthy dog fecal samples used in this study were collected in the spring [15], two dogs (HDS1 and HDS2) were also sampled the previous fall, creating a paired set of samples from the same healthy dogs approximately five months apart. The weighted Unifrac distances between the paired HDS1 and HDS2 samples were 0.255 and 0.208, respectively. These distances were significantly smaller than the average distance between healthy non-enriched profiles (0.404 ± 0.151), indicating that samples from the same animal at different times are more similar to each other than samples taken from different animals at the same time. This result was expected and has been found repeatedly in human microbiome studies [39–41].
Bacteroidetes, and more specifically Bacteroides species, were significantly reduced in diarrheic dogs relative to healthy dogs in this study (Fig. 1b and Table 3). Bacteroides are among the most abundant organisms isolated from healthy dogs by culture [20, 23] and the most frequently detected in sequence-based studies [18, 19]. A single probe-based (FISH) study has suggested that dogs suffering from chronic diarrhea contain more Bacteroides than healthy animals [24], while a 16S rRNA T-RFLP analysis found that Bacteroides spp. levels decreased in diarrhea [13]. Our results agree with the latter study. The presence of Bacteroides have been linked to intestinal health in several ways, including being associated with a healthy body weight in mice [39] and humans [42] and inducing tolerance to commensal bacteria at the intestinal mucosa, preventing inappropriate inflammatory responses in mice [43].
Fusobacteria have been reported to be a major component of the dog fecal microbiome, detected at levels of 108–9 organisms/g of healthy dog feces by culture [20, 23] and comprising from 14% to 40% of the 16S rRNA sequences detected in some studies [19, 44]. Alternatively, other 16S rRNA studies have detected Fusobacteria at an abundance of <1% of the total microbiota [18]. When the same dog fecal samples were examined by two different methods, 16S rRNA pyrosequencing predicted the prevalence of Fusobacteria to be 27–44% of the microbiome [19], while unamplified whole metagenome sequencing predicted the same group to represent only 7–8% of sequences obtained [17]. Fusobacteria sequences were detected in this study, with four healthy dogs and three diarrheic dogs comprising a total of three OTU. However, the abundance of these sequences were <1% of the total microbiota in these animals. This suggests that Fusobacteria detection may be affected by the methodology used.
Epsilon-proteobacteria, including both Campylobacter and Helicobacter, are readily detected in canine feces by targeted PCR [15, 45]. However, these members of the rare microbiome have not been detected by DGGE, T-RFLP, and 16S rRNA-based methods [13, 14, 18, 19]. It was therefore somewhat unexpected that epsilon-proteobacteria sequences comprised 2.66% of the non-enriched healthy profiles and 0.02% of the diarrheic ones (Fig. 1b, Table 4). This increased detection could be a reflection of the alternative gene target used in this study (cpn60), as Proteobacteria sequences in general were detected at a higher prevalence in this study (∼23%) compared to 16S rRNA-based pyrosequencing studies (<15%) [17–19]. The dominant epsilon-proteobacteria detected in healthy dogs were Campylobacter upsaliensis and Helicobacter canis (Table 4). This agrees with the current understanding of dog fecal microbiota [9, 15, 46]. A Helicobacter sequence distinct from any helicobacter in our reference database was also detected in the libraries (labeled Helicobacter sp.) (Supplementary Figure 6). Unfortunately, as cpn60 sequences for all described Helicobacter species (such as for “H. heilmannii”) are not available at this time, we cannot determine if this Helicobacter is a novel species or a previously described one missing from our reference collection. As well, there is the possibility that the single OTU that was similar to C. gracilis could be a novel Campylobacter species (Table 4; Supplementary Figure 6). The reference sequence collection of cpn60 sequences for the Campylobacter genus is extensive, and a pairwise identity of 89.2% is suggestive of a distinct species in this genus (Supplementary Figure 6).
Molecular enrichment increased the proportion of epsilon-proteobacteria sequences from 2.66% to 81.81% (31-fold increase) in healthy dog profiles and from 0.02% to 23.19% (1,160-fold increase) in diarrheic profiles. With the enrichment, almost all the epsilon-proteobacteria sequences seen in the non-enriched profiles were still detected, but at a higher abundance, and additional OTUs were detected that had not been seen before (Table 4). Non-epsilon-proteobacteria sequences detected in the enriched libraries were predominantly Firmicutes, with very few Bacteroides sequences (Supplementary Table 2). The greatest overall difference in the epsilon-proteobacteria community was seen in the diarrheic profiles. Even though epsilon-proteobacteria only comprised 23% the enriched profiles, five additional species (A. skirrowii, C. concisus, C. jejuni, Flexispira rappini, and H. cinaedi) were detected. This contrasts the healthy dog profiles, where only A. skirrowii was detected by enrichment only (Table 4).
The methods used in this study to perform enrichment for rare bacterial sequences could be transferred to any target gene and any group of interest. The only requirement is for sequence knowledge about a group of interest outside a universal target region. The advantage of studying a rare group of organisms by an enrichment approach, as compared to a targeted PCR approach, is that knowledge of specific target species is not required. This approach also avoids a problem often encountered with species-specific PCR where an assay may be too specific for a particular strain or subset of strains within a species. The detection of four distinct genera within the target class (Arcobacter, Campylobacter, Flexispira, and Helicobacter), including possible novel species, make this a powerful method for rare microbiome investigation. There has been discussion in the literature regarding the presence of the “rare biosphere” in 16S rRNA-based deep sequencing studies that appears as a long tail of singletons in rank abundance curves of OTU and the extent to which it is an artifact of the methodology [1, 2]. While some sequence diversity is undoubtedly introduced by PCR and sequencing, our results clearly demonstrate that enrichment can reveal real microbial diversity at the species level, undetected in the first tier of sequencing effort.
References
Reeder J, Knight R (2009) The ’rare biosphere’: a reality check. Nat Methods 6:636–637
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P (2010) Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12:118–123
Ipsos-Reid (2001) Paws and claws: a syndicated study on Canadian pet ownership. Ipsos-Reid, Toronto, pp 1–176
AVMA (2007) US Pet ownership and demographics sourcebook. American Veterinary Medical Association, Schaumburg
Fullerton KE, Ingram LA, Jones TF, Anderson BJ, McCarthy PV, Hurd S, Shiferaw B, Vugia D, Haubert N, Hayes T, Wedel S, Scallan E, Henao O, Angulo FJ (2007) Sporadic Campylobacter infection in infants: a population-based surveillance case-control study. Pediatr Infect Dis J 26:19–24
Tenkate TD, Stafford RJ (2001) Risk factors for campylobacter infection in infants and young children: a matched case-control study. Epidemiol Infect 127:399–404
Carrique-Mas J, Andersson Y, Hjertqvist M, Svensson A, Torner A, Giesecke J (2005) Risk factors for domestic sporadic campylobacteriosis among young children in Sweden. Scand J Infect Dis 37:101–110
Tsai HJ, Huang HC, Lin CM, Lien YY, Chou CH (2007) Salmonellae and campylobacters in household and stray dogs in northern Taiwan. Vet Res Commun 31:931–939
Rossi M, Hanninen ML, Revez J, Hannula M, Zanoni RG (2008) Occurrence and species level diagnostics of Campylobacter spp., enteric Helicobacter spp. and Anaerobiospirillum spp. in healthy and diarrheic dogs and cats. Vet Microbiol 129:304–314
Engvall EO, Brandstrom B, Andersson L, Baverud V, Trowald-Wigh G, Englund L (2003) Isolation and identification of thermophilic Campylobacter species in faecal samples from Swedish dogs. Scand J Infect Dis 35:713–718
Hald B, Pedersen K, Waino M, Jorgensen JC, Madsen M (2004) Longitudinal study of the excretion patterns of thermophilic Campylobacter spp. in young pet dogs in Denmark. J Clin Microbiol 42:2003–2012
Koene MG, Houwers DJ, Dijkstra JR, Duim B, Wagenaar JA (2004) Simultaneous presence of multiple Campylobacter species in dogs. J Clin Microbiol 42:819–821
Bell JA, Kopper JJ, Turnbull JA, Barbu NI, Murphy AJ, Mansfield LS (2008) Ecological characterization of the colonic microbiota of normal and diarrheic dogs. Interdiscip Perspect Infect Dis 2008:149694
Gronvøld AM, L’Abée-Lund TM, Sørum H, Skancke E, Yannarell AC, Mackie RI (2010) Changes in fecal microbiota of healthy dogs administered amoxicillin. FEMS Microbiol Ecol 71:313–326
Chaban B, Ngeleka M, Hill JE (2010) Detection and quantification of 14 Campylobacter species in pet dogs reveals an increase in species richness in feces of diarrheic animals. BMC Microbiol 10:73
Suchodolski JS (2011) Microbes and gastrointestinal health of dogs and cats. J Anim Sci 89:1520–1530
Swanson KS, Dowd SE, Suchodolski JS, Middelbos IS, Vester BM, Barry KA, Nelson KE, Torralba M, Henrissat B, Coutinho PM, Cann IK, White BA, Fahey GC Jr (2011) Phylogenetic and gene-centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice. ISME J 5:639–649
Handl S, Dowd SE, Garcia-Mazcorro JF, Steiner JM, Suchodolski JS (2011) Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS Microbiol Ecol 76:301–310
Middelbos IS, Vester Boler BM, Qu A, White BA, Swanson KS, Fahey GC Jr (2010) Phylogenetic characterization of fecal microbial communities of dogs fed diets with or without supplemental dietary fiber using 454 pyrosequencing. PLoS One 5:e9768
Davis CP, Cleven D, Balish E, Yale CE (1977) Bacterial association in the gastrointestinal tract of beagle dogs. Appl Environ Microbiol 34:194–206
Simpson JM, Martineau B, Jones WE, Ballam JM, Mackie RI (2002) Characterization of fecal bacterial populations in canines: effects of age, breed and dietary fiber. Microb Ecol 44:186–197
Greetham HL, Giffard C, Hutson RA, Collins MD, Gibson GR (2002) Bacteriology of the Labrador dog gut: a cultural and genotypic approach. J Appl Microbiol 93:640–646
Mentula S, Harmoinen J, Heikkilä M, Westermarck E, Rautio M, Huovinen P, Könönen E (2005) Comparison between cultured small-intestinal and fecal microbiotas in beagle dogs. Appl Environ Microbiol 71:4169–4175
Jia J, Frantz N, Khoo C, Gibson GR, Rastall RA, McCartney AL (2010) Investigation of the faecal microbiota associated with canine chronic diarrhoea. FEMS Microbiol Ecol 71:304–312
Schellenberg J, Links MG, Hill JE, Dumonceaux TJ, Peters GA, Tyler S, Ball TB, Severini A, Plummer FA (2009) Pyrosequencing of the chaperonin-60 universal target as a tool for determining microbial community composition. Appl Environ Microbiol 75:2889–2898
Hill JE, Town JR, Hemmingsen SM (2006) Improved template representation in cpn60 polymerase chain reaction (PCR) product libraries generated from complex templates by application of a specific mixture of PCR primers. Environ Microbiol 8:741–746
Schellenberg J, Links MG, Hill JE, Hemmingsen SM, Peters GA, Dumonceaux TJ (2011) Pyrosequencing of chaperonin-60 (cpn60) amplicons as a means of determining microbial community composition. Methods Mol Biol 733:143–158
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Hill JE, Penny SL, Crowell KG, Goh SH, Hemmingsen SM (2004) cpnDB: a chaperonin sequence database. Genome Res 14:1669–1675
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235
Lozupone C, Hamady M, Knight R (2006) UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7:371
Felsenstein J (1989) PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 5:164–166
Sakamoto M, Suzuki N, Benno Y (2010) hsp60 and 16S rRNA gene sequence relationships among species of the genus Bacteroides with the finding that Bacteroides suis and Bacteroides tectus are heterotypic synonyms of Bacteroides pyogenes. Int J Syst Evol Microbiol 60:2984–2990
Desai AR, Musil KM, Carr AP, Hill JE (2009) Characterization and quantification of feline fecal microbiota using cpn60 sequence-based methods and investigation of animal-to-animal variation in microbial population structure. Vet Microbiol 137:120–128
Mansfield GS, Desai AR, Nilson SA, Van Kessel AG, Drew MD, Hill JE (2010) Characterization of rainbow trout (Oncorhynchus mykiss) intestinal microbiota and inflammatory marker gene expression in a recirculating aquaculture system. Aquaculture 307:95–104
Dumonceaux TJ, Hill JE, Hemmingsen SM, Van Kessel AG (2006) Characterization of intestinal microbiota and response to dietary virginiamycin supplementation in the broiler chicken. Appl Environ Microbiol 72:2815–2823
Hill JE, Fernando WM, Zello GA, Tyler RT, Dahl WJ, Van Kessel AG (2010) Improvement of the representation of bifidobacteria in fecal microbiota metagenomic libraries by application of the cpn60 universal primer cocktail. Appl Environ Microbiol 76:4550–4552
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI (2009) A core gut microbiome in obese and lean twins. Nature 457:480–484
Fierer N, Lauber CL, Zhou N, McDonald D, Costello EK, Knight R (2010) Forensic identification using skin bacterial communities. Proc Natl Acad Sci USA 107:6477–6481
Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R (2009) Bacterial community variation in human body habitats across space and time. Science 326:1694–1697
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023
Round JL, Mazmanian SK (2010) Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA 107:12204–12209
Suchodolski JS, Camacho J, Steiner JM (2008) Analysis of bacterial diversity in the canine duodenum, jejunum, ileum, and colon by comparative 16S rRNA gene analysis. FEMS Microbiol Ecol 66:567–578
Haesebrouck F, Pasmans F, Flahou B, Chiers K, Baele M, Meyns T, Decostere A, Ducatelle R (2009) Gastric helicobacters in domestic animals and nonhuman primates and their significance for human health. Clin Microbiol Rev 22:202–223
Harbour S, Sutton P (2008) Immunogenicity and pathogenicity of Helicobacter infections of veterinary animals. Vet Immunol Immunopathol 122:191–203
Acknowledgments
This study was funded by the WCVM Companion Animal Health Fund and the Saskatchewan Health Research Foundation. BC was supported by a postdoctoral fellowship from the Saskatchewan Health Research Foundation. The authors are grateful to Manuel Chirino for donating the genomic DNA used in epsilon-enrichment primer development.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chaban, B., Links, M.G. & Hill, J.E. A Molecular Enrichment Strategy Based on cpn60 for Detection of Epsilon-Proteobacteria in the Dog Fecal Microbiome. Microb Ecol 63, 348–357 (2012). https://doi.org/10.1007/s00248-011-9931-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-011-9931-7