
Abstract
Tricholoma scalpturatum is an ectomycorrhizal fungus that forms symbioses with roots of diverse trees and shrubs. It is commonly encountered in a wide range of habitats, across temperate ecosystems. A previous study has revealed a high genetic diversity at a local scale, and ruderal abilities. To examine genetic structure at a large geographical scale, a total of 164 basidiocarps were collected from 30 populations located in Western Europe, from Spain to Scandinavia. These samples were analyzed by three molecular methods with different levels of resolution: inter-simple sequence repeats (ISSRs), restriction fragment length polymorphisms (RFLPs) in the rDNA internal transcribed spacer (ITS), and ITS sequence analysis. Considerable genetic variation was found, and the morphospecies was separated into two genetic groups that were distinct from each other. The ISSR data and the relatively low percentage value (96%) of shared sequence polymorphisms in the ITS between isolates from the two groups, strongly suggest cryptic species and long-lasting separation. No geographical exclusion was detected for these two widely distributed taxa. However, high estimates of population differentiation were observed in each group, including between populations less than a few kilometers apart. This result provides evidence for limited gene flow and/or founding effects. It also indicates that T. scalpturatum does not constitute a random mating population, and the hypothesis of endemism cannot be excluded for this cosmopolitan wind-dispersed fungus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Numerically, the Fungi constitute a major group of Eukaryota. There are vast numbers of species—a conservative estimate is 1.8 million species [22]—but about 70,000 (i.e., less than 5%) have been described to date. Among these, a great number of morphospecies (i.e., a species definition based on phenotypic descriptions) have broad geographical distribution, covering up to several continents. However, little is known about the dispersal abilities of fungi relatively to those of macroorganisms because of difficulties in observing individuals. For instance, global perspective on fungal populations is relatively sparse [25] and has been mostly investigated on human agent diseases (e.g., Cryptococcus neoformans [48]), plant pathogens (e.g., Mycosphaerella graminicola [50]), and saprotrophic fungi with economical consequences (e.g., Serpula himantioides [28]).
Fungi range in size from microscopic unicells (e.g., yeasts) to vegetative filaments that differentiate macroscopic sexual fruit bodies (known as mushrooms). Within these reproductive structures, meiosis occurs and leads to the production of sexual spores. Regardless of their sexual or vegetative origin, spores are naturally dispersed by wind, water, or animals, potentially over large geographical ranges [6]. Along with vast population sizes, this has contributed to the common view in the mycological literature that endemic species should be rare and population differentiation expected to be minimal. However, there is considerable evidence for cryptic species and/or ecological adaptations in natural fungal populations, suggesting dispersal limitations, recent speciation events, and potential endemism [6]. For example, recent investigations of genetic diversity for the dry rot basidiomycete S. himantioides have led to the recognition of at least three genetically defined cryptic species [28]. In the free-living saprotrophic fungi Neurospora intermedia, distinct ecotypes are correlated with substrate and the geographical area in which they occur [46 in 6]. Similarly, distinct distributions of genotypes are often reported in biotrophic fungi in relation to the host. For example, the rice blast fungus, Magnaporthe grisea, includes several interfertile races that occur on generically distinct graminaceous crops (e.g., rice, foxtail millet, wheat) [6]. Recently, Murat et al. [34] found strong patterns of geographic differentiation in the Perigord black truffle, Tuber melanosporum, over populations from Western Europe. From these data, they suggested that the ectomycorrhizal mycobiont population structure was shaped by the host postglacial range expansion.
Tricholoma scalpturatum (Fr.) Quelet is an ectomycorrhizal basidiomycete fungus symbiotically associated with roots of a broad range of angiosperm and gymnosperm trees and shrubs. This fungus is encountered throughout the Northern hemisphere (e.g., Europe [4, 8], and North America [36]), and is fairly common. It colonizes diverse habitats such as mixed conifer–deciduous forests, riparian forests, or planted trees in city parks. T. scalpturatum is also described as a rather variable species with respect to morphology [4, 8, 36]. To our knowledge, this species has not received attention for potential commercial applications, either in reforestation programs or mushroom production. Therefore, it provides an interesting model to infer contemporary and historical processes of population structure in widely distributed ectomycorrhizal fungi, compared to human-managed species (e.g., Cantharellus spp. [11], Tuber spp. [38], Pisolithus spp. [32]). Recently, Gryta et al. [19] used spatial and temporal mapping of fruit bodies and identification of multilocus genotypes (= genets) to investigate the effect of habitat disturbances on two T. scalpturatum populations from the South of France. Their study revealed a high diversity over small areas, suggesting that sexual reproduction plays an important role in the local occupation of space and ruderal abilities for this fungus. The investigation of relationships among genets placed them in two genetic groups, called groups 1 and 2. The population from the lowest disturbed habitat was found to have individuals from the two groups.
In the present study, the following questions were addressed: (1) what is the genetic diversity of the morphospecies T. scalpturatum (sensu Bon [4]) in Western Europe, and how is it distributed among populations? and (2) are the two genetic groups [19] present at a large geographical scale? Basidiocarps were collected from Southern to Northern Europe in a broad range of habitats. Genetic markers were used to detect polymorphism at different levels of resolution, including sequence comparisons and restriction fragment length polymorphisms (RFLPs) of the nuclear rDNA ITS to delineate genetic groups, and inter-simple sequence repeats (ISSRs) to access genetic relationships among samples.
Materials and Methods
Fungal Sampling and Conservation
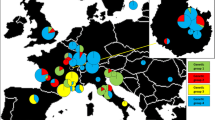
A total of 164 basidiocarps were collected from 30 localities throughout Western Europe, from Spain to Sweden (Fig. 1, Table 1). The samples cover a wide range of habitats across the distribution range of the species and include a broad variety of ectomycorrhizal host trees (Table 1). Collections made by EDB laboratory (Toulouse 3) consisted of a small amount of fresh cap tissues in Eppendorf tubes containing a CTAB buffer (100 mM Tris HCl (pH 8), 1.4 M NaCl, 20 mM Na2 EDTA, and 2% CTAB); the specimens were also air dried. Collections from Denmark and Sweden (Table 1) consisted of dried specimens stored at the Botanical Museum in Copenhagen, Denmark (C).

Sampling location of T. scalpturatum basidiocarps. Full circles represent the 30 local populations and full lines the seven regions. Number of samples (n) and genets (ng) within each local population, region, and Northern vs Southern of Europe are presented. Asterisks indicate local populations that contained genets from groups 1 and 2
Isolation of Genomic DNA
Genomic DNA was extracted from inner caps and gills using the Wizard genomic DNA purification kit (Promega). Extraction was carried out following the manufacturer’s protocol with minor modifications. The fungal material was transferred to a microcentrifuge tube containing 500 μl of the Nuclei Lysis Solution (Promega), and ground using a pestle disposable mixer (Dutscher, France). The resulting homogenate was incubated at 65°C for 15 min, after which 20 μl of RNase A (10 mg/ml) were added to the solution, and the sample incubated at 37°C for 15 min. After incubation, the Protein Precipitation Solution (200 μl) was added to the tube and the mixture centrifuged at 13 000 rpm for 10 min. After isopropanol precipitation and washing with 70% ethanol, the air-dried DNA’s pellet was resuspended in 40 μl of sterile deionized water. Concentration was estimated by electrophoresis in a 0.8% agarose gel stained with ethidium bromide and calibration against known amounts of bacteriophage λ DNA markers under UV illumination with the help of Bio1D++ software (Vilber Lourmat, France). Before amplification, each DNA extract was diluted to 10 ng/μl.
Sequencing and RFLP Analysis of the Internal Transcribed Spacer Region
Fourteen basidiocarps representative of the two genetic groups revealed by ISSR analysis (see Results) were chosen for sequence comparison of the internal transcribed spacer (ITS) region. Amplification was carried out using the primer pair ITS1-F/ITS4-B [17] in a 25-μl reaction volume containing: approximately 10–15 ng of template DNA, 0.2 mM deoxyribonucleotide triphosphate each, 1 μM of each primer, 1 U of Taq DNA polymerase (Go Taq™, Promega) together with the buffer recommended by the manufacturer (5× Go Taq™ Reaction Buffer). Cycling parameters included an initial denaturation at 94°C for 3 min followed by 35 cycles at 94°C for 45 s, annealing at 55°C for 45 s, and 72°C for 1 min, with a final extension at 72°C for 10 min, after which the reaction was cooled to a constant 4°C. The polymerase chain reaction (PCR) products were commercially sequenced in both directions (GenomExpress, Grenoble, France), with ITS1 and ITS4 [49] as sequencing primers. All sequences were edited using BioEdit Version 5.0.9 [20] and aligned using Clustal X version 1.82 [44] with manual correction. Sequences were submitted to GenBank (Table 2).
Based on the nucleotidic variation identified by ITS sequence analysis (cf. Results), the endonuclease Tru9I (Promega) was chosen to study restriction fragment length polymorphism (RFLP) among the 164 basidiocarps of the entire population. ITS amplification was carried out as described above, and the PCR product verified on agarose gels stained with ethidium bromide. The amplified DNA was digested for 3 h at 65°C with Tru9I, according to the manufacturer’s recommendations (Promega). Restriction fragments were electrophoretically separated on 2% composite agarose gels (standard agarose: NuSieve agarose (FMC Bioproduct) 1:1), stained with ethidium bromide, viewed under UV light and digitally photographed using a CDD camera monitored by BioCapt Software (Vilber Lourmat, France). Fragment sizes were calibrated against a 100-bp ladder (Promega). Correspondence between the ITS-RFLP patterns (= Tru9I ribotypes) and the ISSR patterns (see below) was examined for the 164 studied basidiocarps.
ISSR Patterns
A total of nine ISSR primers were screened for polymorphisms against a subset of 12 basidiocarps collected from different sites. The optimal annealing temperature was determined for each primer independently, using the Eppendorf Mastercycler Gradient thermocycler. From the nine primers, four were selected based on easy to interpret patterns and the presence of polymorphic fragments (Table 3).
The PCR reaction was as described above except for the primer concentration (2 μM). The amplification procedure included an initial denaturation at 94°C for 3 min followed by 37 cycles at 94°C for 45 s, annealing at a temperature dependent of the primer (between 50°C and 61°C) for 45 s, and 72°C for 3 min, with a final extension at 72°C for 10 min, after which the reaction was cooled to a constant 4°C. For all the samples, reactions were carried out using the same thermocycler. Negative controls (without template DNA) were included in every run to test for contamination in the reagents. Amplification products were separated by electrophoresis in 1.6% agarose gels and photographed under UV light as described above.
Band scoring and sizing were carried out using the BIO 1D++ V97.06 software (Vilber Lourmat, France) and independently performed by two persons. Well-separated bands and bands that were shared by at least two samples were subsequently considered for the establishment of a binary matrix of presence (1)/absence (0).
To assess the reproducibility of the ISSR assays, PCR reactions were repeated at least twice for half of the samples, and DNA from 12 selected basidiocarps were re-extracted and re-assayed at various times. The effect of material conservation on the ISSR patterns was tested on a subset of samples by comparing fresh and dried specimens. To facilitate inter-gel comparisons, positive controls (= DNA from one to ten selected samples) were included at each PCR run and electrophoresis.
Analyses of Genetic Diversity and Spatial Population Structure
Nei and Li’s genetic distance [35] was calculated among all pairs of basidiocarps, as \({\text{GD}} = 1 - {{2n_{xy} } \mathord{\left/ {\vphantom {{2n_{xy} } {\left( {n_x + n_y } \right)}}} \right. \kern-\nulldelimiterspace} {\left( {n_x + n_y } \right)}}\) where n x and n y refers to the number of ISSR bands that were present in sample X and Y, respectively, and n xy to the number of ISSR bands shared by the two samples. Genetic similarity index (SI) between sample pairs was calculated from GD values as \({\text{SI}} = 1 - {\text{GD}}\) [35]. Basidiocarps for which SI = 1 were considered of the same genet. An UPGMA dendrogram was drawn using the genetic distance matrix with the software TREECON 1.3b [47] to represent the genetic relationships between the studied samples. The confidence of the UPGMA representation was tested with 100 bootstraps. To assess whether any group structure could be detected among genets, principal coordinate analysis (PCA) was computed in PCO [1] using the estimated pairwise genetic distances between genets.
To test for spatial population structure, genets were grouped in a hierarchical manner based on their geographical positions on a map: (1) local population (= groups of genets separated from each other by less than 2 km), (2) regional population (= groups of genets within a given region), (3) Northern population (= Scandinavian genets from Denmark and Sweden), and (4) Southern population (= group of genets from France and Spain). Thus, a total of 30 local populations were identified, and each was assigned to one of the following regions: Sweden (I), Denmark (II), Alps (III), Corsica island (IV), Mediterranean border (V), Midi-Pyrénées (i.e., Southwest of France with Atlantic influences) (VI), and Massif Central (VII; Table 1, Fig. 1). Ecogeographical parameters, climate, geology, and hydrology were used to define the seven regions. Analysis of molecular variance (AMOVA, [12]) was carried out using ARLEQUIN 2.0 software package [39] to test for the distribution of genetic variability: (1) among and within local populations, (2) among and within regional populations, and (3) among and within the Northern and Southern populations (Fig. 1). Population differentiation among local populations was assessed from AMOVA using the index ΦST, analogous to Wright’s F ST.
The Mantel test [31] was used to estimate the correlation between genetic distance, as estimated by ΦST, and the geographical distances (in km) between populations. The test was implemented in ADE-4 software package [43] with 1000 random permutations for estimation of significance.
Results
Genetic Diversity Within the Morphospecies T. scalpturatum
Sequence analysis obtained from 14 basidiocarps yielded 27 variable sites in the amplified 695 bp sequence of the ribosomal ITS (Table 2). Twenty-four of these sites were located outside known endonuclease restriction sites, but three fell into the TTAA region of the Tru9I enzyme (Table 2). This characteristic allowed rapid screening of genetic variation after digestion of the amplified ITS and scoring of fragments on agarose electrophoresis gels. Among all basidiocarps analyzed (n = 164), only two Tru9I RFLP patterns (referred to as ribotypes 1 and 2) were identified across all geographic locations. From BioEdit calculating, ribotype 1 was characterized by fragments of 317, 261, 88, and 23 bp in size, whereas ribotype 2 had fragments of 267, 243, 162, and 23 bp. Differences in fragment sizes between the two profiles were due to site mutations. Ribotype 1 was encountered more frequently among the analyzed basidiocarps: 91 (55%) and 73 (45%) basidiocarps had ribotype 1 and ribotype 2, respectively.
ISSR amplifications performed on 164 basidiocarp DNAs using four primers yielded 73 clear and reproducible bands, of which all were polymorphic (Table 3). Separate amplification produced uniform fragments, and no difference was seen between fresh and dry material from selected basidiocarps. Fingerprints consisted in 15–21 bands depending on the primer, ranging in size from 252 to 1580 bp (not shown). The combined data sets resulted in 147 multilocus genets (Fig. 2), of which 137 (93%) were unique and 10 (7%) represented by two to seven site-specific basidiocarps. The average value (%) of genetic distance (GD) between genet pairs was 73.6 (range from 2.7 to 100).

Despite relatively low bootstrap values, the UPGMA cluster analysis based on calculated GDs showed that T. scalpturatum comprised two distinct ISSR clusters (GD = 0.95), with 88 and 59 genets from various geographical locations in group 1 (G1) and group 2 (G2), respectively (Fig. 2). The principal component analysis revealed a similar genetic structure, with the first and second component accounting for 45.6% and 10.3% of the total variance, respectively (Fig. 3). Thirty-seven bands (out of 73; 51%) were shared between the two genetic clusters, whereas 13 (18%) and 23 (31%) were encountered only in G1 and G2, respectively (not shown). Within each cluster population, the mean percentage value of GD was 52.5 and 57 for genets of group 1 (range 3.2–85.7) and group 2 (range 2.7–100), respectively. The mean similarity index (SI) between pairs of genets was also calculated for groups 1 and 2. The highest value was obtained for group 1 (0.477±0.139, frequency range from 0.14 to 0.96), compared to group 2 (0.422±0.140, frequency range from 0 to 0.97) (Fig. 4). Of interest, low values of similarity index were obtained between the two groups (mean = 0.074 ± 0.076, frequency range from 0 to 0.44; Fig. 4), suggesting the existence of two genetically isolated groups. Last, AMOVA also showed a high and significant level of differentiation between the two groups (Φst = 0.41, P < 0.001).

Principal coordinate analysis of the 147 genets of Tricholoma scalpturatum based on ISSR data. Genets of group 1 or 2 as described by UPGMA cluster analysis are encompassed by dashed lines. The different symbols refer to the regions where the genets originated from

Frequency distribution of similarity index calculated among genet pairs within group 1 (in black), within group 2 (in white), and between groups 1 and 2 (in grey). The means of the similarity index are indicated at the top of the figure
Comparison of the data obtained from Tru9I ribotyping, ITS sequencing, and ISSR fingerprinting showed a perfect match in the identification of two genetic groups within the morphospecies T. scalpturatum. Thus, ribotype 1 included all the basidiocarps from ISSR cluster 1, whereas ribotype 2 comprised ISSR cluster 2 basidiocarps. The aligned sequences also emphasized the two groupings within T. scalpturatum. Basidiocarps from the same ISSR genetic cluster had 100% sequence homology with each other, but only 96.1% homology with basidiocarps from the other cluster.
Population Differentiation and Spatial Structure
The two ribotypes were widespread throughout Europe, from Spain to Sweden (Fig. 1, Table 1). No relationship was found between the dominant plant host and the presence of either Tru9I ribotype 1 or 2. Both were found under species of conifers or deciduous trees (Table 1). Finally, the two groups were observed sympatrically in five populations from France (III.14, III.15, III.16, V.21, and VI.25; Fig. 1, Table 1).
To investigate spatial structure, ISSR genets were grouped according to geographical position (i.e., localities, regions, North vs South), and analyses of molecular variance were performed to test the significance of these groups. When population genets from both genetic clusters were pooled, AMOVA showed that 55.9% of the genetic variance observed was found within local populations and 44.1% among populations, indicating a very high and significant level of population differentiation (Φst = 0.44, P < 0.001; Table 4). However, there were concerns of obtaining high Φst values due to sampling effect. Indeed, if local populations belonged by chance to either genetic group 1 or 2, this could generate differences due not to the genetic difference between populations but between genetic groups. Low sample sizes in most local populations were also another potential source of bias in the Φst estimation. To test these effects, AMOVA was carried out within each genetic group independently, and Φst calculated for populations of low (local) to large (regions, North and South populations) sample sizes. Data still showed very high and significant levels of genetic differentiation between local populations in each group (Φst = 0.31 and 0.49 in groups 1 and 2, respectively; P < 0.001; Table 4). Most of the genetic variance observed was within populations (68.9% and 50.6% in groups 1 and 2, respectively). Similarly, when AMOVA was performed for regional populations and Northern vs Southern populations, the differentiation indexes were high and significant (Table 4). For instance, at the regional scale, Φst was 0.21 and 0.28 with P < 0.001 for groups 1 and 2, respectively. The largest amount of variation was explained by the differences within regional populations (79.3% and 72.0% for groups 1 and 2, respectively).
Based on the AMOVA results that suggested differentiation between populations, a Mantel test was performed to examine correlations between geographic distance and genetic distance. Under this model, more distantly separated populations are expected to exhibit higher differentiation if there is lower gene flow. Group 1 always showed high and significant correlations for populations at local (Mantel r = 0.19, P = 0.019) and regional (r = 0.51, P = 0.024) scales. However, the relationship was not significant for group 2 (not shown), indicating no distinct pattern of isolation by distance at these two geographical scales.
Discussion
The discovery of previously unappreciated diversity in organisms displaying morphological stasis, e.g., fungi, is often reported [e.g., 10, 28, 30, 37]. Our ISSR results supports the recognition of two distinct clusters within the morphospecies T. scalpturatum [19]. ITS polymorphism also demonstrates the existence of two groups that were consistent with the two ISSR clusters. When all data are considered (ISSR, ITS-RFLPs and sequences), the morphospecies T. scalpturatum is clearly separated in two genetic groups, called genetic groups 1 and 2, which are assumed to be closely related species between which reproductive isolation may be complete. However, with respect to these data, a reliable phylogenetic framework based on multigene datasets [9, 41] is a prerequisite for addressing questions about evolution and speciation in T. scalpturatum.
The utility of the ITS for delimiting species is well documented in fungal systematics [e.g., 5, 45]. For instance, it is often claimed that two isolates must show a similarity >97% to be considered part of the same ectomycorrhizal species [e.g., 24, 40, 42]. Out of the 14 ITS sequences, we consistently found 3.9% of sequence divergence between isolates from the two genetic groups of T. scalpturatum and no intra-group polymorphism. The ITS region is also extensively used in molecular ecology of ectomycorrhizal fungi to identify taxonomic units from environmental samples (mycorrhizae and soil) [3, 23]. As more sequences will be available in GenBank, the accuracy of identification will improve [29], and fast methods of identification (e.g., DNA barcoding and phylogenetic microarrays for community studies) could developed on the basis of sequence data [3].
The two genetic groups (thereafter also referred to as cryptic species) have a wide range distribution within Western Europe, from Spain to Scandinavia. This pattern is consistent with the traditional view of cosmopolitan fungi [37] and microorganism ubiquity [13, 14, 16]. A widespread distribution of cryptic species has also been observed in the human pathogenic fungus Aspergillus fumigatus sampled from worldwide locations [37]. However, many fungi postulated to have large repartition (intercontinental or continental) often contain narrowly distributed genetic groups. For example, the cosmopolitan ectomycorrhizal mushroom, Pisolithus tinctorius, is a complex of cryptic species that exhibit geographical patterns in relation to the distribution of their host trees [33]. In the two human fungal pathogens, Histoplasma capsulatum and Coccidioides immitis, biogeographically significant variation was also observed among the phylogenetically identified species [27, 30]. In addition to a widespread distribution, results from T. scalpturatum indicate that the two cryptic species were not geographically exclusive. Thus, genotypes from the two species co-occurred in several populations within restricted areas. Our proposed biogeographical scenario is that the observed diversity is the result of ancient speciation events, reproductive isolation, and secondary contacts. Phylogenetically based taxonomy would provide help to assess the evolutionary relationships between these two codistributed taxa. Our results also raise questions about ecological adaptation within T. scalpturatum. At present, it is not known whether the two cryptic species occupy different ecological niches or frequently grow together on roots. Superficially, fungi may show similar or overlapping ranges, but they often occupy microhabitats such as adjacent sites on a single log or different humus layers in soils [42]. Further evidence is required regarding functional diversity and fine-scale distribution of the two species of T. scalpturatum.
The ability possessed by a large number of fungal species to disperse their spores by wind and potentially exploit a wide range of habitats may perhaps account, in part, for their large geographical distribution. Presumably, genetic crosses between populations should also occur. However, there are many examples of fungal populations that resemble those of other eukaryotes in tending to exhibit population differentiation, associated with different areas, or habitats [6]. Do both genetic groups of T. scalpturatum maintain a global population or are they more likely to show a restricted geographical distribution? Within both groups, high genetic differentiations were observed for local and large (i.e., regions and Northern vs Southern Europe) populations. Like T. scalpturatum, the epigeous ectomycorrhizal basidiomycetes Russula brevipes and Suillus tomentosus showed strong genetic differentiation (Fst = 0.434 and Gst = 0.289, respectively) [2, 51 in 26]. This pattern was tentatively explained by the existence of geographic barriers to dispersal [2, 26], although the existence of cryptic species was also suspected [2]. In animal-dispersed ectomycorrhizal fungi, e.g., T. melanosporum and Rhizopogon, population differentiation was found at small geographical scales, and it was attributed to specialization to the host, habitat fragmentation, geographic barriers, and reduced dispersal abilities [18, 34].
Genetic differentiation among group 1 populations increased with the distance between these populations, but the Mantel test failed to reveal a similar trend in group 2 populations. Reasons may be that the two genetic groups differ in their colonization capabilities, overall densities, and sensitivity to habitat fragmentation. Furthermore, several processes of population evolution have to be taken into account to explain the observed patterns of genetic differentiation. For instance, founded populations may rapidly differentiate from other populations because of limited genetic variability. A small initial population size should also increase the impact of genetic drift and thus the divergence between populations. In addition to low dispersal, the founding effect could therefore play an important role in population differentiation of T. scalpturatum.
Conclusion
When the genetic diversity of T. scalpturatum was investigated using the methods outlined here, it became clear that this morphospecies exhibited considerable variation and was separable into two distinct groups. Our sampling led us to the conclusion that these two genetic groups should be regarded as widespread cryptic species with overlapping geographic ranges. Highly structured populations were also observed over short distances, suggesting barriers to gene flow in each group. The absence of globally mixed populations could therefore support the hypothesis of endemism in wind-dispersed microorganisms [7, 15].
References
Anderson MJ (2003) PCO: a FORTRAN computer program for principal coordinate analysis. Department of Statistics, University of Auckland, New Zealand
Bergemann SE, Miller SL (2002) Size, distribution, and persistence of genets in local populations of the late stage ectomycorrhizal basidiomycete, Russula brevipes. New Phytol 156:313–320
Bidartondo M, Gardes M (2005) Fungal diversity in molecular terms: profiling, identification, and quantification in the environment. In: Dighton J, White TF, Oudemans P (eds) The Fungal Community, 3rd edn. CRC Press, Boca Raton, pp 215–239
Bon M (1984) Les Tricholomes de France et de l’Europe Occidentale. Lechevalier edn., Paris
Bruns TD, White TJ, Taylor JW (1991) Fungal molecular systematics. Annu Rev Ecol Evol Syst 22:525–564
Burnett J (2003) Fungal population and species. Oxford University Press, Oxford, UK
Cho J-C, Tiedje JM (2000) Biogeography and degree of endemicity of fluorescent Pseudomonas strains in soil. Appl Environ Microbiol 60:1227–1234
Christensen M, Noordeloos ME (1999) Notulae ad floram agaricinam neerlandicam-XXXVI Tricholoma. Persoonia 17:295–317
Dettman JR, Jacobson DJ, Taylor JW (2003) A multilocus genealogical approach to phylogenetic species recognition in the model Eukaryote Neurospora. Evolution 57:2703–2720
Douhan GW, Rizzo DM (2005) Phylogenetic divergence in a local population of the ectomycorrhizal fungus Cenococcum geophilum. New Phytol 166:263–271
Dunham SM, Kretzer A, Pfrender ME (2003) Characterization of pacific golden chanterelle (Cantharellus formosus) genet size using co-dominant microsatellite markers. Mol Ecol 12:1607–1618
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Finlay BJ (2002) Global dispersal of free-living microbial eukaryote species. Science 296:1061
Finlay BJ, Clarke KJ (1999) Ubiquitous dispersal of microbial species. Nature 400:828
Fisher MC, Koenig GL, White TJ, San-Blas G, Negroni R, Gutierrez Alvarez I, Wanke B, Taylor JW (2001) Biogeographic range expansion into South America by Coccidioides immitis mirror new world patterns of human migration. Proc Natl Acad Sci USA 98:4558–4562
Garcia-Pichel F, Prufert-Bedout L, Muyer G (1996) Phenotypic and phylogenetic analyses show Microcoleus chthonoplastes to be a cosmopolitan cyanobacterium. Appl Environ Microbiol 62:3284–3291
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118
Grubisha LC, Bergemann SE, Bruns TD (2007) Host islands within the California Northern Channel Islands create fine-scale genetic structure in the two sympatric species of the symbiotic ectomycorrhizal fungus Rhizopogon. Mol Ecol 16:1811–1822
Gryta H, Carriconde F, Charcosset J-Y, Jargeat P, Gardes M (2006) Population dynamics of the ectomycorrhizal fungal species Tricholoma populinum and Tricholoma scalpturatum associated with black poplar under differing environmental conditions. Environ Microbiol 8:773–786
Hall TA (1999) BioEdit software, version 5.0.9. North Carolina State University, Raleigh, NC
Hantula J, Dusabenygasani M, Hamelin RC (1996) Random amplified microsatellites (RAMS)—a novel method for characterizing genetic variation within fungi. Eur J For Path 26:159–166
Hawksworth DL (2001) The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res 105:1422–1432
Horton TR, Bruns TD (2001) The molecular revolution in ectomycorrhizal ecology: peeking into the black-box. Mol Ecol 10:1855–1871
Izzo A, Agbowo J, Bruns TD (2005) Detection of plot-level changes in ectomycorrhizal communities across years in a old-growth mixed-conifer forest. New Phytol 166:619–630
Jacobson DJ, Dettman JR, Adams RI, Boesl C, Sultana S, Roenneberg T, Merrow M, Duarte M, Marques I, Ushakova A, Carneiro P, Videira A, Navarro-Sampedro L, Olmedo M, Corrochano LM, Taylor JW (2006) New findings of Neurospora in Europe and comparisons of diversity in temperate climates on continental scales. Mycologia 98:550–559
James TY, Porter D, Hamrick JL, Vilgalys R (1999) Evidence for limited intercontinental gene flow in the cosmopolitan mushroom, Schizophyllum commune. Evolution 53:1665–1667
Kasuga T, White TJ, Koenig G, Mcewen J, Restrepo A, Castaneda E, Da Silva Lacaz C, Heins-Vaccari EM, De Freitas R, Zancopé-Oliviera RM, Qin Z, Negroni R, Carter DA, Mikami Y, Tamura M, Taylor ML, Miller GF, Poonwan N, Taylor JW (2003) Phylogeography of the fungal pathogen Histoplasma capsulatum. Mol Ecol 12:3383–3401
Kauserud H, Stensrud Ø, DeCock C, Shalchian-Tabrizi K, Schumacher T (2006) Multiple gene genealogies and AFLPs suggest cryptic speciation and long-distance dispersal in the basidiomycete Serpula himantioides (Boletales). Mol Ecol 15:421–431
Kõljalg U, Larsson K-H, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AFS, Tedersoo L, Vrålstad T, Ursing BM (2005) UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol 166:1063–1068
Koufopanou V, Burt A, Szaro T, Taylor JW (2001) Gene genealogies, cryptic species, and molecular evolution in the human pathogen Coccidioides immitis and relatives (Ascomycota, Onygenales). Mol Biol Evol 18:1246–1258
Mantel NA (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Martin F, Delaruelle C, Ivory M (1998) Genetic variability in intergenic spacers of ribosomal DNA in Pisolithus isolates associated with pine, eucalyptus and Aflezia in lowland Kenyan forests. New Phytol 139:341–352
Martin F, Dìez J, Dell B, Delaruelle C (2002) Phylogeography of the ectomycorrhizal Pisolithus species as inferred from nuclear ribosomal DNA ITS sequences. New Phytol 153:345–357
Murat C, Diez LP, Delaruelle C, Dupré C, Chevalier G, Bonfante P, Martin F (2004) Polymorphism at the ribosomal DNA ITS and its relation to postglacial re-colonization of the Perigord truffle Tuber melanosporum. New Phytol 164:401–411
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Ovrebo CL (1989) Tricholoma, subgenus Tricholoma, section Albidogrisea: North American species found principally in the Great Lakes region. Can J Bot 67:3134–3152
Pringle A, Baker DM, Platt JL, Wares JP, Latgé JP, Taylor JW (2005) Cryptic speciation in the cosmopolitan and clonal human pathogenic fungus Aspergillus fumigatus. Evolution 59:1886–1899
Rubini A, Paolocci F, Riccioni C, Vendramin GG, Arcioni S (2005) Genetic and phylogeographic structures of the symbiotic fungus Tuber magnatum. Appl Environ Microb 71:6584–6589
Schneider S, Roessli D, Excoffier L (2000) ARLEQUIN ver 2.000. A software for population genetics data analysis. URL: http:/anthro.unige.ch/arlequin
Smith ME, Douhan GW, Rizzo DM (2007) Ectomycorrhizal community structure in a xeric Quercus woodland based on rDNA sequence analysis of sporocarps and pooled roots. New Phytol 174:847–863
Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31:21–32
Tedersoo L, Kõljalg U, Hallenberg N, Larsson K-H (2003) Fine scale distribution of ectomycorrhizal fungi and roots across substrate layers including coarse woody debris in a mixed forest. New Phytol 159:153–165
Thioulouse J, Chessel D, Dolédec S, Olivier J-M (1997) ADE-4: a multivariable analysis and graphical display software. Stat Comput 7:75–83
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24:4876–4882
Tsui CKM, Sivichai S, Berbee ML (2006) Molecular systematics of Helicoma, Helicomyces and Helicosporium and their teleomorphs inferred from rDNA sequences. Mycologia 98:94–104
Turner B (1987) Two ecotypes of Neurospora intermedia. Mycologia 79:425–432
Van de Peer Y, De Wachter R (1994) TREECON for windows: a software package for construction and drawing of evolutionary trees for Microsoft Windows environment. Comp Appl Bio sci 10:569–570
Viviani MA, Cogliati M, Esposto MC, Lemmer K, Tintelnot K, Colom Valiente MF, Swinne D, Velegraki A, Velho R, Group ECoMMECW (2006) Molecular analysis of 311 Cryptococcus neoformans isolates from 30-month ECMM survey of cryptococcosis in Europe. FEMS Yeast Res 6:614–619
White TJ, Bruns TD, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for polygenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, New York, USA, pp 315–322
Zhan J, Pettway RE, McDonald BA (2003) The global genetic structure of the wheat pathogen Mycosphaerella graminicola is characterized by high nuclear diversity, low mitochondrial diversity, regular recombination, and gene flow. Fungal Genet Biol 38:286–297
Zhu H, Higginbotham KO, Dancik BP (1988) Intraspecific genetic variability of isozymes in the ectomycorrhizal fungus Suillus tomentosus. Can J Bot 66:588–594
Acknowledgments
We thank Dr. Darin Penneys and Dr. Wieland Meyer for critical review of the manuscript and Dr. Lounès Chikhi for advices and suggestions during the course of the project. We also gratefully acknowledge Dr. Pierre-Arthur Moreau, Jacques Fournier, Marie Fournier, and Pr. Guy Durrieu for assistance with collecting, Sophie Manzi and Nicolas Paoli for technical assistance in the molecular analysis. Funding for this research was provided by a MENRT grant “ACI Microbiologie” to M.G.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Carriconde, F., Gardes, M., Jargeat, P. et al. Population Evidence of Cryptic Species and Geographical Structure in the Cosmopolitan Ectomycorrhizal Fungus, Tricholoma scalpturatum . Microb Ecol 56, 513–524 (2008). https://doi.org/10.1007/s00248-008-9370-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-008-9370-2