
Abstract
Tuber species are ectomycorrhizal ascomycetes establishing relationships with different host trees and forming hypogeous fruiting bodies known as truffles. Among Tuber species, Tuber aestivum Vittad. has a wide distributional range being found naturally all over Europe. Here, we performed large-scale population genetic analyses in T. aestivum to (i) investigate its genetic diversity at the European scale, (ii) characterize its genetic structure and test for the presence of ecotypes and (iii) shed light into its demographic history. To reach these goals, 230 ascocarps from different populations were genotyped using 15 polymorphic simple sequence repeat markers. We identified 181 multilocus genotypes and four genetic groups which did not show a clear geographical separation; although, one of them was present exclusively in Southeast France, Italy and Spain. Fixation index values between pairs of genetic groups were generally high and ranged from 0.29 to 0.45. A significant deficit of heterozygosity indicated a population expansion instead of a recent population bottleneck, suggesting that T. aestivum is not endangered in Europe, not even in Mediterranean regions. Our study based on a large-scale population genetic analysis suggests that genetically distinct populations and likely ecotypes within T. aestivum are present. In turn, this study paves the way to future investigations aimed at addressing the biological and/or ecological factors that have concurred in shaping the population genetic structure of this species. Present results should also have implications for the truffle market since defining genetic markers are now possible at least for some specific T. aestivum genetic groups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The ectomycorrhizal (ECM) symbiosis is a worldwide mutualistic association between fungi and plant roots (Smith and Read 2008). ECM fungi provide nutrients to plants and therefore play a key role in their nutrition. Moreover, intraspecific mycorrhizal diversity can influence plant diversity, as well as ecosystem functioning (Johnson et al. 2012). Knowledge on the genetic diversity and population genetic structure of a species is indispensable to understand its biology and demography. Due to the development of polymorphic molecular markers, population genetic studies of ECM fungi are now more feasible and are increasing in numbers (Douhan et al. 2011). On the one hand, large-scale population genetic studies have shown the presence of isolated sub-populations in some ectomycorrhizal species such as Laccaria amethystina (Hortal et al. 2012), Rhizopogon roseolus (Okuda et al. 2013), Tuber brumale (Merényi et al. 2014), Tuber magnatum (Rubini et al. 2005) and Tuber melanosporum (Murat et al. 2004; Riccioni et al. 2008; García-Cunchillos et al. 2014). On the other hand, the absence of any genetic differentiation among populations was shown in Pisolithus microcarpus (Hitchcock et al. 2011) and Suillus spraguei (Rivera et al. 2014).
The population genetic structure of a species, corresponding to the distribution of genotypes in populations, results from a combination of its propagation modes and demographic processes. For ECM fungi, several spore propagation modes exist such as wind for epigeous species and animals for hypogeous species. The propagation modes can have a significant effect on gene flow, and epigeous fungi such as Russula brevipes exhibit strong genetic differentiation between populations at large scale but not at smaller scale (Bergemann et al. 2006). On the contrary, for hypogeous fungi such as Rhizopogon and Tuber melanosporum, much greater population differentiation at smaller spatial scales was observed (Murat et al. 2004; Grubisha et al. 2007).
Truffles are hypogeous ascomycetes, which belong to the Tuber genus found over the entire Northern hemisphere. More than 200 Tuber species have been reported worldwide (Bonito et al. 2010, 2013), and to date, 32 truffle species have been described in Europe (Ceruti et al. 2003), many of which are precious edible fungi. As ECM fungi, truffles play an important role in woodland ecosystems specifically for tree growth (Smith and Read 2008). Methods to obtain plant roots mycorrhized by edible truffles including T. aestivum and T. melanosporum have been already established in the 1970s (Murat 2015). Under natural conditions, some truffle species, such as T. magnatum and T. melanosporum, show specific and limited geographical distribution. T. magnatum is a species harvested only in Italy and in some countries on the Balkan Peninsula, with a few samples found in the South of France; T. melanosporum is naturally found mainly in France, Italy and Spain. However, other species have a broader distribution. For example, T. aestivum is found all over Europe, from Sweden to Spain, and has also been observed in North Africa (Jeandroz et al. 2008). Different ecological requirements, genetic diversities and/or demographic histories might cause such variations in distribution.
Tuber aestivum, described for the first time by Carlo Vittadini in 1831 (Vittadini 1831), presents a black peridium and a brown gleba (Montecchi and Sarasini 2000; Callot 1999; Chevalier and Frochot 2002). This species shows an ecological polymorphism with different maturity periods and ecological preferences depending on its geographical location (Chevalier et al. 1979; Chevalier and Frochot 2002; Riousset et al. 2001; Stobbe et al. 2013). In 1887, Chatin split T. aestivum into two distinct species: T. aestivum and Tuber uncinatum on the basis of spore morphological characteristics (Chatin 1887). Since that time, T. aestivum and T. uncinatum have either been considered as a single species or as two different species depending on the authors (Dupré et al. 1985; Gandeboeuf et al. 1994; Mello et al. 2002; Mouches et al. 1981; Pacioni et al. 1993; Pacioni and Pomponi 1991; Paolocci et al. 2004; Urbanelli et al. 1998; Weden et al. 2005). Recent phylogenetic analyses suggest that existing morphological differences between T. aestivum and T. uncinatum do not reflect two different species but rather suggest that they should be considered as a single species: T. aestivum (Paolocci et al. 2004; Weden et al. 2005; Molinier et al. 2013b). Nevertheless, Le Tacon (2011) proposed the presence of ecotypes within T. aestivum due to the high genetic diversity observed in this species associated with its large-scale distribution in Europe. The concept of ecotype was introduced by Turesson 1922: ‘The term ecotype is proposed here as [an] ecological unit to cover the product arising as a result of the genotypical response of an ecospecies to a particular habitat’. Ecotypes are therefore populations or sub-units of a species that are adapted to a particular environment. In summary, the T. aestivum/T. uncinatum controversy highlights, in a more general context, the question about the species concept in fungal species (Taylor et al. 2000).
The genetic structure in Europe and gene dispersion of T. aestivum are not fully addressed yet. Among Tuber spp. these analyses have mainly been carried out in T. melanosporum, T. magnatum and T. brumale (Murat et al. 2004, 2011, 2013; Rubini et al. 2004; Riccioni et al. 2008; Merényi et al. 2014; García-Cunchillos et al. 2014). For example, a strong genetic structure with significant fixation indices was observed among T. melanosporum populations at a European scale (F st = 0.20 and F st = 0.177; Murat et al. 2004; Riccioni et al. 2008). Concerning T. aestivum, previous studies mainly tested the hypothesis of a genetic differentiation between T. aestivum and T. uncinatum morphotypes, not least because commercially relevant, and were based on a limited sample size and/or low number of molecular markers (Mello et al. 2002; Paolocci et al. 2004; Weden et al. 2005; Molinier et al. 2013b). Splivallo et al. (2012) investigated the genetic diversity of T. aestivum from nine European populations by amplified fragment length polymorphism, but they focused on the comparison between genetic and volatile profiles. Interestingly, the samples coming from the same populations tended to cluster together, but due to the low number of samples analysed (i.e. 42), it was not possible to draw a definitive conclusion on the genetic structure of T. aestivum populations at the European scale. At small scale, by investigating the genetic structure of T. aestivum in two orchards in France, Molinier et al. (2015) reported that the size of the genets found was higher with respect to the size of T. melanosporum genets recorded in two black truffle orchards (Murat et al. 2013). The T. aestivum population genetic structure at European scale is therefore an issue that remains to be investigated.
On this concern, present study was focused on T. aestivum specimens genotyped with 15 polymorphic simple sequence repeat (SSR) markers recently characterized in this species (Molinier et al. 2013a) with the aims to (i) investigate the genetic diversity at European scale, (ii) test for the presence of genetic groups and (iii) shed light into the demographic history (of this species). We hypothesized that (i) a geographical population structuration exists over Europe and (ii) ecotypes of this species are present. Besides being biologically relevant, characterizing the genetic diversity and determining ecotype existence in T. aestivum has applied relevance for the truffle industry. It can help selecting well-adapted inoculum for the production of mycorrhizal seedlings to increase the cultivation success of this species.
Materials and methods
Sampling and DNA extraction
Two hundred and thirty ascocarps of Tuber aestivum (fruiting bodies) were collected from natural populations in 12 European countries (Table 1). The samples analysed here are available upon request. Each sample was crushed using a sterilized scalpel. DNA was extracted by the Plateforme Gentyane, INRA Clermont-Ferrand (France). Briefly, an extraction buffer (100 mM Tris pH = 8.0, 50 mM EDTA, 5 M NaCl, 20 mM metabisulfite and 2 % RNase (10 mg/ml)) was added to the crushed truffles, and tubes were incubated at 95 °C for 45 min. Tubes were then centrifuged for 10 min at 4000g (4 °C). Supernatants were then transferred into new tubes containing isopropanol/ammonium acetate (7.5 M) (400/25, v/v) and homogenized by inversion. Tubes were centrifuged during 30 min at 4000g at 4 °C. Supernatants were discarded, and pellets were washed once with 70 % Ethanol. After drying, the pellets were resuspended in 100 μL of Tris EDTA 0.1×. DNA concentration was estimated using PicoGreen®, and all DNA extracts were adjusted to 10 ng/ml.
PCR amplification and genotyping
Fifteen polymorphic microsatellite loci previously defined (Molinier et al. 2013a) were used (aest1, aest7, aest6, aest10, aest15, aest18, aest24, aest25, aest26, aest27, aest28 aest29, aest31, aest35 and aest36). Microsatellite polymerase chain reactions (PCR) were performed in a 10-μL volume containing 2.5 μl at 10 ng/ml of DNA of T. aestivum, 5 μL of AmpliTaq Gold® 360 Master Mix (AB-life technologies), 0.5 μL primer mix fluorescently labelled forward primer with either 6-FAM, VIC (Eurofins MWG Operon) fluorescent dyes (Molinier et al. 2013a) and 2 μL sterile water. PCR was performed in a thermocycler Veriti 384 Well (Applied Biosystems) under the following conditions: the PCR started with a cycle of 10 min at 95 °C, followed by 7 cycles of 30 s at 95 °C, 30 s at 62 °C and 30 s at 72 °C. The PCR further continued with 30 cycles of 30 s at 95 °C, 30 s at 55 °C and 30 s at 72 °C and after 8 cycles of 30 s at 95 °C, 30 s at 56 °C and 30 s at 72 °C and finished with an elongation cycle of 5 min at 72 °C. PCR products were assayed on an ABI 3730XL sequencer (Applied Biosystems, Foster City, CA, USA) using GENESCANTM 500 liz as a size standard. Fragment sizes were analysed with GENEMAPPER 3.7 (Applied Biosystems).
Genetic analyses
Genetic diversity and identification of multilocus genotypes
The DNA extracted from truffle ascocarps with classical extraction protocols is mainly contributed by the gleba which is formed by a haploid tissue (Paolocci et al. 2006). All tests realized in this study are suitable for haploid organisms. For each SSR marker, the number of alleles (Na), number of effective alleles (Ne), expected heterozygosity (He) and allelic frequencies were calculated using GenAlex version 6.501 (Peakall and Smouse 2006). The expected heterozygosity measures the probability that two copies of the same gene chosen randomly in a population will have different alleles (Nei 1973). The number of multilocus genotypes (MLGs) and genotypic diversity alone, as well as versus the number of loci, were calculated using Multilocus 1.3b (Agapow and Burt 2001). The genotypic diversity is the probability that two individuals taken at random have different genotypes. In other words, this value is 0 if every individual has the same genotype and 1 if every individual has a different genotype.
When different samples showed the same MLG, the MLGsim software (Stenberg et al. 2003) was used to distinguish true clones from samples carrying the same multilocus genotype by chance. MLGsim computed the probability of observing a multilocus genotype n times in a population by chance (Psex), and Monte Carlo simulations (10,000 iterations) were performed to determine the significance of this probability.
Genetic structure analyses
The occurrence of population subdivision was investigated using the Bayesian clustering method implemented in GENELAND (Guillot 2008; Guillot et al. 2005b). GENELAND infers the best number of genetic groups (K). The number of genetic groups that best explain the data is given by the value of K in which the Markov chain spent most iterations (Guillot et al. 2005a; Guillot et al. 2005b). The number of K was determined by running the MCMC iterations 10 times, allowing K to vary from 1 to 10 with the following parameters: 550,000 iterations combined with the uncorrelated frequency model and not using the spatial model. A burn-in of 100 saved iterations (i.e. discarding the 100 first saved iterations) was applied in post-processing. For GENELAND, analyses have been performed with MLG data to avoid statistical bias.
Multilocus and pairwise linkage disequilibrium (LD) analyses were performed using MULTILOCUS 1.3b over all MLG and per genetic groups. To evaluate the LD, the rBarD index (Agapow and Burt 2001) was calculated in each case. The same software was used to determine if rBarD differed significantly from zero, by performing 1000 randomizations in each case. First, the randomization procedure was performed by shuffling alleles among all individuals (MLG). To determine if LD results from a genetic structure, randomizations by shuffling alleles among individuals within genetic groups identified by GENELAND were also performed.
Genetic group pairwise Fst calculations, pairwise kinship coefficients (Fij) and intra genetic group kinship coefficients (Gij) were calculated using SPAGeDi software (Hardy and Vekemans 2002). The interrelation among MLGs was analyzed by correspondence analysis and visualized according to their group membership using the ADE4 package of R (http://www.r-project.org/) (Dray and Dufour 2007; Thioulouse et al. 1997; Thioulouse and Dray 2007).
Deviation from mutation-drift equilibrium
The deviation from mutation-drift equilibrium in the genetic groups was tested using BOTTLENECK software version 1.2.02 (Piry et al. 1999). The heterozygosity deficit may provide evidence for population expansion while the heterozygosity excess may provide evidence for a genetic bottleneck (Cornuet and Luikart 1996; Luikart et al. 1998a, b). Using a Wilcoxon test, the heterozygosity deficit and heterozygosity excess have been evaluated under three mutation models: two-phase mutation model (TPM), infinite allele model (IAM) and stepwise mutation model (SMM) (Cornuet and Luikart 1996; Di Rienzo et al. 1994). The Wilcoxon test was chosen since it is preferable when few loci are used (Cornuet and Luikart 1996; Cornuet et al. 1999). The TPM model was used with 12 % variation from the IAM model and 95 % from the SMM model as recommended for microsatellites (Piry et al. 1999).
A second approach for bottleneck assessment was to test a mode shift away from an L-shaped distribution of allelic frequencies (Luikart et al. 1998a, b). Using GeneAlex v 6.41, the allele frequency of all loci for each genetic group was calculated. Then, a graphical representation of the proportion of alleles in the different allele frequency was realized to check for an L shape. Indeed, a population that experienced a bottleneck is unlikely to exhibit an L-shaped distribution of allelic frequencies.
Results
Genetic diversity at the European scale
A total of 230 ascocarps of Tuber aestivum harvested in Europe were successfully genotyped using 15 SSR markers (Table 1). As expected, due to the haploid nature of the gleba (Paolocci et al. 2006), each locus resulted in one peak for each sample. All SSRs were polymorphic and presented a number of alleles ranging from 5 to 24 (Table S1).
Since the SSRs used were not linked, as per linkage disequilibrium analyses reported below, all the markers were kept, and by combining the allelic profiles at the 15 SSR loci, a total of 181 MLGs were identified (Table S2). When we plotted the genotypic diversity and the number of MLGs against the number of loci, a plateau was observed with a genotypic diversity of 0.99 for five loci, although the number of genotypes continued to increase up to 15 loci (Fig. S1). MLGsim analysis (Stenberg et al. 2003) confirmed that the samples showing the same MLG were likely to belong to the same genet, and thus, they were regarded to as true clones; the PSex index was in fact lower than 0.01 (Table S2). Out of the 26 clonal MLG identified, nine comprised samples from different geographical origins, as far apart as the UK and Belgium (Table S2).
Identification of T. aestivum genetic groups and demographical history
When all 181 MLG were included, the multilocus analysis had high significant departure from linkage equilibrium (rBarD = 0.15, p value < 0.001; Table S3). Similarly, the two-locus analysis had significant LD between almost all pairs of loci. The rBarD values decreased significantly when they were calculated within the four genetic groups (see below). Similarly, no pair of loci exhibited significant departure from linkage equilibrium within the four genetic groups. These data suggest that our markers were not genetically linked (Table S3).
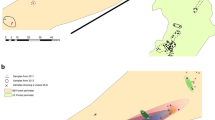
Within the entire T. aestivum collection, GENELAND analysis identified four different genetic groups with the highest posterior probabilities (Fig. S2; Table 2). The four genetic groups did not correspond to specific geographical areas (Fig. 1). The fixation index (Fst) values between pairs of genetic groups ranged from 0.29 to 0.45 (Table 3). For each genetic group, the expected heterozygosity (He), reflecting the genetic diversity inside the group, was high but not noticeably different across groups (Table 2). High kinship coefficient (Fij) values, corresponding to an estimator of genetic relatedness, were observed when pairs of samples within the same genetic group were considered, while lower and negative kinship values were observed between samples from different groups (Table 3).

Geographical distribution of the four genetic groups defined using GENELAND. For each location, the proportion of samples belonging to genetic groups 1 (green), 2 (red), 3 (yellow) and 4 (blue) is indicated. The size of the pie is proportional to the number of samples
Assignation as ‘T. aestivum’ or ‘T. uncinatum’ morphotype was conducted for each sample according to morphological criteria as described by Chevalier and collaborators (Chevalier et al. 1979; Table S4). Interestingly, the genetic group 3 turned out to contain exclusively samples morphologically classified as T. aestivum (Table S4). Correspondence analysis supported the group differentiation as revealed by GENELAND analyses and indicated a slight differentiation between T. uncinatum and T. aestivum morphotypes (Fig. 2a, b). When superposing Fig. 2a, b, it appeared that mainly samples belonging to the T. aestivum morphotype and corresponding to the genetic group 3 were isolated from T. uncinatum ones (Fig. 2).

Correspondence analysis showing the relation among samples based on their MLG with samples visualized according to their genetic group membership (on the left; a) and pre-assignation (on the right;; b)
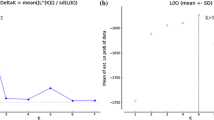
A significant excess of heterozygosity was detected only for genetic group 4 under assumption of the IAM model to suggest a recent bottleneck (Table 4). On the contrary, a significant heterozygosity deficit was detected for all genetic groups with both SSM and TPM models suggesting a population expansion (Table 4). The discrepancy between the three model tests was probably caused by different expected heterozygosities at mutation equilibrium (Luikart and Cornuet 1998). In addition, all four genetic groups displayed an L-shaped distribution of allele frequencies excluding the presence of a recent bottleneck (Fig. 3).

Distribution of allele frequencies expected for loci evolving under the infinite allele model of mutation (IAM) in the four genetic groups at mutation-drift equilibrium. Black bars represent the proportion of alleles expected in each of the 11 allele frequency classes
Discussion
In this study, we examined at a large scale the genetic structure of Tuber aestivum. To reach this end, samples from populations harvested in 12 different European countries were investigated using 15 polymorphic SSRs. Our main goal was to unveil more on the genetic pattern of this species and identify potential genetic groups. Overall, the presence of four genetic groups not corresponding to geographical groups was disclosed as well as the concomitant presence within the same populations of different well-differentiated genetic groups. These results also suggest the presence of ecotypes within this species.
T. aestivum has a high genetic diversity at the European scale and probably a long-distance dispersion
Over the 230 samples, the number of alleles identified ranged from 5 to 24. This resembles the number of SSR alleles found in T. magnatum (2 to 18 alleles on 315 samples; Rubini et al. 2004) and T. melanosporum (2 to 18 alleles on 139 samples; Murat et al. 2011). The average He of 0.50 (Table 2) that we detected in T. aestivum is the highest ever found in a truffle species. At large scale, lower genetic diversity levels were in fact found in T. mesentericum (He = 0.21; Pomarico et al. 2007), T. borchii (He = 0.17; Pomarico et al. 2007), T. magnatum (He = 0.16; Pomarico et al. 2007), T. brumale fo. moschatum (He = 0.09; Pomarico et al. 2007) and T. melanosporum (He = 0.41; Murat et al. 2011). Our values are similar and sometimes even higher than He values found in air-dispersed ECM fungi such as Laccaria species (He = 0.41/0.46; Wadud et al. 2014) or Suillus spraguei (He = 0.33; Rivera et al. 2014). T. aestivum is therefore a species with a high genetic diversity at a European scale.
As regards the distribution of the MLG throughout our sampling area, some samples with identical MLG were as far apart as the UK, Belgium and Sweden (Table S2). At first glance, this suggests a spread of genets throughout Europe. This phenomenon was previously observed in some air-dispersed fungi such as Laccaria amethystina (Hortal et al. 2012; Vincenot et al. 2012) or Tricholoma scalpturatum (Carriconde et al. 2008 and Jargeat et al. 2010). Hypogeous fungi are conversely animal dispersed, and thus, it is surprising to find a spread of truffle genets throughout Europe. However, as highlighted by Vellinga et al. (2009), humans are moving fungi across landscapes. Due to its organoleptic qualities and since inoculated seedlings with T. aestivum are available since 40 years (Murat 2015), humans might have probably contributed to spread truffles throughout Europe. The use of additional molecular markers is essential to test whether or not T. aestivum strains are truly shared between different countries.
Within T. aestivum genetically distinct groups and likely ecotypes are present
Within the four genetic groups identified by GENELAND, both two locus and multilocus linkage disequilibrium analyses indicate that extensive genetic exchange occurs. However, the evidence that some samples from the same population belong to different genetic groups suggests that some individuals from the same location would not cross. Since geographical barriers within each location are unlikely to be present, the absence of crossing between individuals from the same population could be explained by ecological differences and/or genetic barriers. We note that the coexistence of different genetic groups with no geographical barriers preventing gene flow has already been observed in L. amethystina (Hortal et al. 2012; Vincenot et al. 2012). Due to the secrecy of truffle harvesting, we have no precise data on our sampling sites to address if and which ecological differences could explain the isolation of samples within the same population. Concerning putative genetic barriers, we note that truffles are heterothallic species and that the mating type genes have been recently identified and characterized in T. melanosporum and T. indicum (Rubini et al. 2011; Belfiori et al. 2013). The genome of T. aestivum is currently undergoing sequencing (Payen et al. 2014). Our results highlight the importance to characterize the mating type genes in T. aestivum samples belonging to different genetic groups to assess whether differences in the sequence and organization of these genes could explain their isolation.
Furthermore, although the genetic groups identified in T. aestivum show an overlapping geographical distribution, it is interesting to note that group 3 is restricted to Spain, Italy and south-eastern France in Europe (Fig. 1). In addition to this, this group is the most differentiated one when compared to the others (Table 3) and only shows the T. aestivum morphotype (Fig. 2). These results let us argue that the genetic group 3 might represent a cryptic species or ecotype within T. aestivum and that it is potentially adapted to southern climate. Ecotypes have been already characterized in other ECM fungi. Suillus species evolved ecotypes that are specifically tolerant against high Zn, Cd or Cu concentrations (Adriaensen et al. 2005; Colpaert et al. 2000), and Pisolithus albus populations from ultramafic soils of New Caledonia are genetically structured in ecotypes (Jourand et al. 2010). The existence of ecotypes within T. aestivum has already been suggested but has not been demonstrated until now (Le Tacon 2011). According to truffle growers, T. aestivum sensu stricto has a distributional range limited to open woodlands located in the southern European regions; it can form a truffle burn area (known as “brulé”) all around its host species, and its fruiting bodies can be harvested in the summer. Contrarily, T. aestivum sensu lato can be found throughout Europe in more mature forests, forms small or no truffle burn and produces fruiting bodies throughout the year (Stobbe et al. 2012). As pointed out earlier, a dedicated study will be needed to unveil whether genetic barriers or differences in ecological specializations might explain not only the presence of distinct genetic groups but also that of possible ecotypes in T. aestivum. Up to now, some private alleles not found in other genetic groups do exist in genetic group 3, which could likely represent genetic markers to identify T. aestivum sensu stricto.
Species and ecotypes discrimination in T. aestivum is an important issue relevant to both science and economy. Indeed, several national laws exist to control truffle commercialization in different countries (e.g. law n°752 December 16th 1985 in Italy; decree n°2012-129 January 30th 2012 in France). Key to this legislation is the ability to precisely define species that can be sold, notably avoiding multiple nomenclatures of the same species which could lead to misinterpretation.
T. aestivum is not an endangered species
Each Tuber spp. is harvested in different environments ranging from managed plantations (particularly for T. melanosporum) to natural forests (specifically for T. magnatum and T. aestivum). However, T. aestivum shows a higher adaptability to a wide climate range as opposed to T. melanosporum or T. magnatum, being harvested all over Europe. Recently, it has been proposed that truffles are endangered in Mediterranean regions and are moving to northern populations due to climate change (Büntgen et al. 2012). In order to test a recent bottleneck of T. aestivum, which would indicate a population contraction putatively due to climate change, we performed two tests: the first based on heterozygosity deficit and the second on the L-shaped distribution of allele frequencies. A significant heterozygosity deficit was detected for all genetic groups with both SSM and TPM models suggesting a population expansion (Table 4). Although a deficit of heterozygosity could also be due to sub-population structure, a recent bottleneck could be excluded as confirmed by L-shaped distribution of allele frequencies. The absence of a recent bottleneck suggested that T. aestivum is not yet an endangered species, not even in Mediterranean regions since genetic group 3 is limited to southern populations (see above). A similar result was found for T. melanosporum in the Iberian Peninsula indicating a non-threatened genetic conservation status (García-Cunchillos et al. 2014). However, even under conservative scenarios, future climate changes are likely to cause further increases in mean temperature (about 2–4 °C globally) with significant drought in some regions (Seager et al. 2007) as well as increases in frequency and severity of extreme droughts, extreme heat levels and heat waves (IPCC 2013; Sterl et al. 2008). Since we know that climate, mainly water deficit, has an effect on truffle fructification (Le Tacon et al. 2014), impacts on truffle populations in the future cannot be excluded as suggested by Büntgen et al. (2012). Therefore, the high level of genetic diversity found as well as its genetic structure with putative presence of ecotypes are key aspects that need to be considered for future T. aestivum cultivation programmes. A recent study carried out in T. melanosporum based on genome re-sequencing of six isolates from different geographical areas has allowed the identification of single nucleotides and putative genes under selection and demonstrated that population genomics can be applied in truffle species to identify genetic traits related to adaptation to environmental conditions (Payen et al. 2015). Likewise, a genome sequencing and re-sequencing project of T. aestivum samples from different areas and belonging to different genetic groups will allow to address the adaptation capacities of this species and verify if ecotypes adapted to particular environments exist within this species.
References
Adriaensen K, Vrålstad T, Noben JP, Vangronsveld J, Colpaert JV (2005) Copper-adapted Suillus luteus, a symbiotic solution for pines colonizing Cu mine spoils. Appl Environ Microbiol 71:7279–7284
Agapow PM, Burt A (2001) Indices of multilocus linkage disequilibrium. Mol Ecol Notes 1:101–102
Belfiori B, Riccioni C, Paolocci F, Rubini A (2013) Mating type locus of Chinese black truffles reveals heterothallism and the presence of cryptic species within the T. indicum species complex. PLoS One 8, e82353
Bergemann SE, Douhan GW, Garbelotto M, Miller SL (2006) No evidence of population structure across three isolated subpopulations of Russula brevipes in an oak/pine woodland. New Phytol 170:177–184
Bonito GM, Gryganskyi AP, Trappe JM, Vilgalys R (2010) A global meta-analysis of Tuber ITS rDNA sequences: species diversity, host associations and long-distance dispersal. Mol Ecol 19:4994–5008
Bonito G, Smith ME, Nowak M, Healy RA, Guevara G, Cázares E et al (2013) Historical biogeography and diversification of truffles in the Tuberaceae and their newly identified southern hemisphere sister lineage. PLoS One 8, e52765
Büntgen U, Egli S, Camarero JJ, Fischer EM, Stobbe U, Kauserud H, Tegel W, Sproll L, Stenseth NC (2012) Drought-induced decline in Mediterranean truffle harvest. Nat Clim Chang 2:827–829
Callot G (1999) La Truffe, La Terre, la Vie. INRA, Paris, 210p
Carriconde F, Gardes M, Jargeat P, Heilmann-Clausen J, Mouhamadou B, Gryta H (2008) Population evidence of cryptic species and geographical structure in the cosmopolitan ectomycorrhizal fungus, Tricholoma scalpturatum. Microb Ecol 56:513–524
Ceruti A, Fontana A, Nosenzo C (2003) Le specie Europee del genere Tuber, una revisione storica. Museo Regionale di Scienze Naturali, Monographie XXXVII, Regione Piemonte, Torino
Chatin A (1887) Une nouvelle espèce de truffe. C R Acad Sci 104:1132–1135
Chevalier G, Frochot H (2002) La truffe de Bourgogne. Editions Pétrarque, Levallois-Perret, France
Chevalier G, Desmas C, Frochot H, Riousset L (1979) L’ espèce Tuber aestivum Vitt. : I. Définition. Mushroom Science X, (Part 1):957–975
Colpaert JV, Vandenkoornhuyse P, Adriaensen K, Vangronsveld J (2000) Genetic variation and heavy metal tolerance in the ectomycorrhizal basidiomycete Suillus luteus. New Phytol 147:367–379
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M (1999) New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics 153:1989–2000
Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci U S A 91:3166–3170
Douhan GW, Vincenot L, Gryta H, Selosse MA (2011) Population genetics of ectomycorrhizal fungi: from current knowledge to emerging directions. Fungal Biol 115:569–597
Dray S, Dufour AB (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22:1–20
Dupré C, Chevalier G, Branlard G (1985) Caractérisation des Tuber par électrophorèse de leurs protéines. In CR 1er Colloque national sur les technologies de purification des protéines. DPIC-INPL, Paris, pp 465–467
Gandeboeuf D, Dupré C, Chevalier G (1994) Use of isoenzyme analysis to differentiate truffles from Europe. Acta Bot Gallica 141:455–463
García-Cunchillos I, Sánchez S, Barriuso JJ, Pérez-Collazos E (2014) Population genetics of the westernmost distribution of the glaciations-surviving black truffle Tuber melanosporum. Mycorrhiza. doi:10.1007/s00572-013-0540-9
Grubisha LC, Bergemann SE, Bruns TD (2007) Host islands within the California Northern Channel Islands create fine-scale genetic structure in two sympatric species of the symbiotic ectomycorrhizal fungus Rhizopogon. Mol Ecol 16:1811–1822
Guillot G (2008) Inference of structure in subdivided populations at low levels of genetic differentiation—the correlated allele frequencies model revisited. Bioinformatics 24:2222–2228
Guillot G, Mortier F, Estoup A (2005a) GENELAND: a computer package for landscape genetics. Mol Ecol Notes 5:712–715
Guillot G, Estoup A, Mortier F, Cosson JF (2005b) A spatial statistical model for landscape genetics. Genetics 170:1261–1280
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hitchcock CJ, Chambers SM, Cairney JWG (2011) Genetic population structure of the ectomycorrhizal fungus Pisolithus microcarpus suggests high gene flow in south-eastern Australia. Mycorrhiza 21:131–137
Hortal S, Trocha LK, Murat C, Chybicki IJ, Buée M, Trojankiewicz M et al (2012) Beech roots are simultaneously colonized by multiple genets of the ectomycorrhizal fungus Laccaria amethystina clustered in two genetic groups. Mol Ecol 21:2116–2129
IPCC (2013) Fifth Assessment Report Climate Change 2013: The Physical Science Basis—Summary for Policymakers, Working Group I Contribution to the IPCC, 2013
Jargeat P, Martos F, Carriconde F, Gryta H, Moreau PA, Gardes M (2010) Phylogenetic species delimitation in ectomycorrhizal fungi and implications for barcoding: the case of the Tricholoma scalpturatum complex (Basidiomycota). Mol Ecol 19:5216–5230
Jeandroz S, Murat C, Wang YJ, Bonfante P, Le Tacon F (2008) Molecular phylogeny and historical biogeography of the genus Tuber, the ‘true truffles’. J Biogeogr 35:815–829
Johnson D, Martin F, Cairney JWG, Anderson IC (2012) The importance of individuals: intraspecific diversity on mycorrhizal plants and fungi in ecosystems. New Phytol 194:614–628
Jourand P, Ducousso M, Reid R, Majorel C, Richert C, Riss J, Lebrun M (2010) Nickel-tolerant ectomycorrhizal Pisolithus albus ultramafic ecotype isolated from nickel mines in New Caledonia strongly enhance growth of a host plant at toxic nickel concentrations. Tree Physiol 30:1311–1319
Le Tacon F (2011) Point de vue sur Tuber aestivum Vittad. Le Trufficulteur 74:13–17
Le Tacon F, Marçais B, Courvoisier M, Murat C, Becker M (2014) Climatic variations explain annual fluctuations in French Périgord black truffle wholesale markets but does not explain the decrease in black truffle production over the last 48 years. Mycorrhiza 24(1 supplement):115–125
Luikart G, Cornuet JM (1998) Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12:228–237
Luikart G, Allendorf FW, Cornuet JM, Sherwin WB (1998a) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89:238–247
Luikart G, Sherwin WB, Steele B, Allendorf FW (1998b) Usefulness of molecular markers for detecting population bottlenecks and monitoring genetic change. Mol Ecol 7:963–974
Mello A, Cantisani A, Vizzini A, Bonfante P (2002) Genetic variability of Tuber uncinatum and its relatedness to other black truffles. Environ Microbiol 4:584–594
Merényi Z, Varga T, Geml J, Orczán Ákos K, Chevalier G, Bratek Z (2014) Phylogeny and phylogeography of Tuber brumale aggr. Mycorrhiza. doi:10.1007/s00572-014-0566-7
Molinier V, Murat C, Morin E, Gollotte A, Wipf D, Martin F (2013a) First identification of polymorphic microsatellite markers in the Burgundy truffle, Tuber aestivum (Tuberaceae). App Plant Sci 1:1200220
Molinier V, van Tuinen D, Chevalier G, Gollotte A, Wipf D, Redecker D (2013b) A multigene phylogeny demonstrates that Tuber aestivum and Tuber uncinatum are conspecific. Org Divers Evol 13:503–512
Molinier V, Murat C, Frochot H, Wipf D, Splivallo R (2015) Fine-scale spatial genetic structure analysis of the black truffle Tuber aestivum and its link to aroma variability. Environ Microbiol. (accepted) http://onlinelibrary.wiley.com/doi/10.1111/1462-2920.12910/abstract
Montecchi A, Sarasini M (2000) Funghi Ipogei d’Europa. AMB, Brescia Italy
Mouches C, Duthil P, Poitou N, Delmas J, Bove J (1981) Caractérisation des espèces truffières par analyse de leurs protéines en gels de polyacrylamide et application de ces techniques à la taxonomie des champignons. Mushroom Science 11:819–831
Murat C (2015) Forty years of inoculating seedlings with truffle fungi: past and future perspectives. Mycorrhiza 25:77–81
Murat C, Diez J, Luis P, Delaruelle C, Dupre C, Chevalier G et al (2004) Polymorphism at the ribosomal DNA ITS and its relation to postglacial re-colonization routes of the Perigord truffle Tuber melanosporum. New Phytol 164:401–411
Murat C, Riccioni C, Belfiori B, Cichocki N, Labbe J, Morin E et al (2011) Distribution and localization of microsatellites in the Perigord black truffle genome and identification of new molecular markers. Fungal Genet Biol 48:592–601
Murat C, Rubini A, Riccioni C, De la Varga H, Akroume E, Belfiori B et al (2013) Fine scale spatial genetic structure of the black truffle (Tuber melanosporum) investigated with neutral microsatellites and functional mating type genes. New Phytol 199:176–187
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A 70:3321–3323
Okuda Y, Shimomura N, Funato C, Nagasawa E, Matsumoto T (2013) Genetic variation among natural isolates of the ectomycorrhizal hypogenous fungus, Rhizopogon roseolus from Japanese pine forests inferred using AFLP markers. Mycoscience 54:13–18
Pacioni G, Pomponi G (1991) Genotypic patterns of some Italian populations of the Tuber aestivum-Tuber mesentericum complex. Mycotaxon 42:171–179
Pacioni G, Frizzi G, Miranda M, Visca C (1993) Genetics of a Tuber aestivum population (Ascomycotina, Tubérales). Mycotaxon 47:93–100
Paolocci F, Rubini A, Riccioni C, Topini F, Arcioni S (2004) Tuber aestivum and Tuber uncinatum: two morphotypes or two species? FEMS Microbiol Lett 235:109–115
Paolocci F, Rubini A, Riccioni C, Arcioni S (2006) Reevaluation of the life cycle of Tuber magnatum. Appl Environ Microbiol 72:2390–2393
Payen T, Murat C, Bonito G (2014) Truffle phylogenomics: new insights into truffle evolution and truffle life cycle. In Francis M. Martin editor Adv Bot Res Vol. 70, Oxford: United Kingdom, 211–234
Payen T, Murat C, Gigant A, Morin E, De Mita S, Martin F (2015) A survey of genome-wide single nucleotide polymorphisms through genome re-sequencing in the Périgord black truffle (Tuber melanosporum Vittad.). Mol Ecol Resources (in press)
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Piry S, Luikart G, Cornuet JM (1999) Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Hered 90:502–503
Pomarico M, Figliuolo G, Rana GL (2007) Tuber spp. biodiversity in one of the southernmost European distribution areas. Biodivers Conserv 16:3447–3461
Riccioni C, Belfiori B, Rubini A, Passeri V, Arcioni S, Paolocci F (2008) Tuber melanosporum outcrosses: analysis of the genetic diversity within and among its natural populations under this new scenario. New Phytol 180:466–478
Riousset L, Riousset G, Chevalier G, Bardet MC (2001) Truffes d’Europe et de chine. INRA Editions, Paris, France
Rivera Y, Burchhardt KM, Kretzer AM (2014) Little to no genetic structure in the ectomycorrhizal basidiomycete Suillus spraguei (Syn. S. pictus) across parts of the northeastern USA. Mycorrhiza 24:227–232
Rubini A, Topini F, Riccioni C, Paolocci F, Arcioni S (2004) Isolation and characterization of polymorphic microsatellite loci in white truffle (Tuber magnatum). Mol Ecol Notes 4:116–118
Rubini A, Paolocci F, Riccioni C, Vendramin GG, Arcioni S (2005) Genetic and phylogeographic structures of the symbiotic fungus Tuber magnatum. Appl Environ Microbiol 71:6584–6589
Rubini A, Belfiori B, Riccioni C, Tisserant E, Arcioni S, Martin F, Paolocci F (2011) Isolation and characterization of MAT genes in the symbiotic ascomycete Tuber melanosporum. New Phytol 189:710–722
Seager R, Ting M, Held I, Kushnir Y, Lu J, Vecchi G, Huang HP, Harnik N, Lau NC, Li C et al (2007) Model projections on an imminent transition to a more arid climate in southwestern North America. Science 316:1181–1184
Smith SE, Read DJ (2008) Mycorrhizal symbiosis. Academic, London
Splivallo R, Valdez N, Kirchhoff N, Ona MC, Schmidt JP, Feussner I, Karlovsky P (2012) Intraspecific genotypic variability determines concentrations of key truffle volatiles. New Phytol 194:823–835
Stenberg P, Lundmark M, Saura A (2003) MLGsim: a program for detecting clones using a simulation approach. Mol Ecol Notes 3:329–331
Sterl A, Severijns C, Dijkstra H, Hazeleger W, Oldenborgh G, Broeke M, Burgers G, Hurk B, Leeuwen P, Velthoven P (2008) When can we expect extremely high surface temperatures? Geophys Res Lett. doi:10.1029/2008GL034071
Stobbe U, Büntgen U, Sproll L, Tegel W, Egli S, Fink S (2012) Spatial distribution and ecological variation of re-discovered German truffle habitats. Fungal Ecol 5:591–599
Stobbe U, Egli S, Tegel W, Peter M, Sproll L, Büntgen U (2013) Potential and limitations of Burgundy truffle cultivation. Appl Microbiol Biotechnol 97:5215–5224. doi:10.1007/s00253-013-4956-0
Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31:21–32
Thioulouse J, Dray S (2007) Interactive multivariate data analysis in R with the ade4 and ade4TkGUI packages. J Stat Softw 22:1–14
Thioulouse J, Chessel D, Doledec S, Olivier JM (1997) ADE-4: a multivariate analysis and graphical display software. Stat Comput 7:75–83
Turesson G (1922) The genotypical response of the plant species to the habitat. Hereditas 3:211–350
Urbanelli S, Sallicandro P, De Vito E, Bullini L, Biocca E (1998) Biochemical systematics of some species in the genus Tuber. Mycologia 90:537–546
Vellinga EC, Wolfe BE, Pringle A (2009) Global patterns of ectomycorrhizal introductions. New Phytol 181:960–973
Vincenot L, Nara K, Sthultz C, Labbe J, Dubois MP, Tedersoo L et al (2012) Extensive gene flow over Europe and possible speciation over Eurasia in the ectomycorrhizal basidiomycete Laccaria amethystina complex. Mol Ecol 21:281–299
Vittadini C (1831) Monographia tuberacearum. France
Wadud MA, Nara K, Lian C, Ishida TA, Hogetsu T (2014) Genet dynamics and ecological functions of the pioneer ectomycorrhizal fungi Laccaria amethystina and Laccaria laccata in a volcanic desert on Mount Fuji. Mycorrhiza 24:551–563
Weden C, Danell E, Tibell L (2005) Species recognition in the truffle genus Tuber—the synonyms Tuber aestivum and Tuber uncinatum. Environ Microbiol 7:1535–1546
Acknowledgments
The work presented was supported by the ‘Conseil Régional de Bourgogne (Programme Jeune Chercheur Entrepreneur; Grant 20100112095254682–1)’, the ‘Ernst Göhner Stiftung’ and the Swiss State Secretariat for Education, Research and Innovation (SERI; COST Action FP1203). The UMR1136 is supported by a grant overseen by the French National Research Agency (ANR) as part of the ‘Investissements d'Avenir’ programme (ANR-11-LABX-0002-01, Lab of Excellence ARBRE). The authors thank Denise Stalder, Gérard Chevalier, Henri Frochot, Jean-Claude Ferrand, Mr. Déquéant, Mr. Besson, Willy Tegel and all other truffle growers for providing samples. We are grateful to Falbien Halkett for the help in the population genetic analysis, the choice of the tests and their interpretation. We thank Marie-Lara Bouffaud, François Le Tacon, Francis Martin and Stéphane De Mita for providing constructive advice and helpful discussions. We would like to thank Andrea Rubini, Claudia Riccioni and two anonymous reviewers for helpful comments on the manuscript and Aimée Orsini for English language editing. The authors also thank the strategic plateform ‘GENTYANE’ INRA, Ibisa 2009 and its group leader Mr. Poncet.
Author contributions
V.M., C.M. and D.W. designed the experiment. V.M. and C.M. performed experiment and data analyses. B.M. and H.D.V. provided assistance with DNA extraction, SSR amplification and analysis. V.M. and C.M. wrote the manuscript. M.P., A.G., S.E., B.B., F.P. and D.W. contributed to the writing and provided samples. All co-authors approved the final manuscript.
Conflict of interest
The authors declare that they have no competing interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Virginie Molinier and Claude Murat both authors contributed equally to this work.
Rights and permissions
About this article

Cite this article
Molinier, V., Murat, C., Peter, M. et al. SSR-based identification of genetic groups within European populations of Tuber aestivum Vittad. Mycorrhiza 26, 99–110 (2016). https://doi.org/10.1007/s00572-015-0649-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00572-015-0649-0