
Abstract
As a crucial step in the identification of possible association between bacteria and sponges, we investigated if a unique bacterial population community was consistently associated with the surface of the sponge Mycale adhaerens, irrespective of environmental conditions. The composition of bacterial communities associated with the surface of sponges at three geographically distinctive sites in Hong Kong waters over four seasons was examined by analyzing terminal restriction fragment length polymorphism of the bacterial 16S rRNA genes. Statistical analysis indicated that bacterial communities on inanimate reference surfaces (polystyrene dishes deployed in the close vicinity of the sponge colonies for 7 days) had a relatively high degree of both site and seasonal specificities (R statistics of pairwise comparisons ∼1), which might be attributed to the differences in environmental conditions at different sites and seasons. On the contrary, the sponge-surface-associated bacterial communities from different sites and seasons were hardly distinguishable from each other (lowest R = −0.16) but were rather distinctive from the reference bacterial communities (R ∼ 1), suggesting a highly stable and distinctive bacteria–sponge association irrespective of the environmental conditions. The occurrence of some unique bacterial types in the sponge-surface-associated communities over space and time suggests that the associations are consistent and specific.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Marine sponges (Phylum: Porifera) are sessile organisms that effectively filter out microorganisms and small particles from the surrounding water. Whereas most “captured” microorganisms serve as food particles and are digested by phagocytotically active cells in sponge, the others are retained, even thrive to reach a balanced state, and finally form specific associations with sponges. Therefore, sponge–microorganism associations become a common phenomenon in nature. For instance, unicellular algae [84], cyanobacteria [82], dinoflagellates [32], zooxanthellae [72], zoochlorellae [34, 95], and members of the domain Archaea [29, 65] have been reported as associates in sponges. Among these microorganisms, bacteria are the most dominant group that constituted up to 40% of the biomass or 60% of the tissue volume of certain sponge species [70, 84, 89, 90].
Sponge–bacterium associations promote mutual proliferation of nutrients and digestion and recycling of materials [7, 91]. Furthermore, bacteria in the sponges are the biosynthetic origins of highly diverse secondary metabolites that are potent antibacterial [49], antifeeding [96], and allelopathic [23] compounds. These metabolites provide chemical defense against predation, microbial attachment, and fouling by other invertebrates for the sponges [52]. In addition, sponges serve as a shelter for bacteria against grazers or the substrate for bacteria to attach so that bacteria can proliferate more rapidly because sponge metabolites can serve as a consistent nutrient supply [43]. There has also been evidence supporting the hypothesis that bacteria associated with sponge surfaces help sponge defend against colonization by other harmful organisms [36, 41, 44, 49, 53, 54, 79].
Earlier studies of sponge–bacterium associations mainly relied on morphological descriptions using transmission electron microscopy [84] and numerical taxonomy using cultivation [71]. However, these methods generally suffer from the fact that bacteria are largely indistinguishable by morphological characteristics, and that <1% of bacteria are cultivable from marine sponges [39]. Recent advances in molecular techniques have provided culture-independent tools to reveal the diversity and precise phylogenetic affiliation of bacteria associated with sponges by targeting the 16S rRNA genes in the bacterial community under investigation. A series of remarkable investigations by Friedrich et al., using fluorescence in situ hybridization, denaturing gradient gel electrophoresis (DGGE), and the construction of DNA libraries, have shown that: (1) bacterial community in the Mediterranean sponge Aplysina cavernicola is dominated by bacteria with low guanine–cytosine content, including the delta- and gamma-Proteobacteria and representatives of the Bacteroides cluster [27]; (2) bacterial community in a congeneric sponge Aplysina aerophoba was surprisingly resistant to environmental perturbations [28, 83]; and (3) some bacteria in the phylogenetically distantly related sponges (A. aerophoba and Thenoella swinhoei) in nonoverlapping geographical locations are highly similar [38].
In contrast to the extensive studies on the bacterial community in the mesohyl of sponges for years [28, 38, 83, 84, 90], the description of the bacterial community associated with sponge surfaces is scarce. Moreover, past investigations generally employed a snapshot sampling strategy [57, 71, 92] that often does not allow an insight into the composition of sponge-associated bacterial communities over time. The present study investigated the compositional stability of the bacterial communities associated with the surface of the sponge Mycale adhaerens commonly found in Hong Kong waters. We compared the variations of bacterial community compositions on both the sponge and reference surfaces (i.e., polystyrene dishes deployed in the close vicinity of the sponge colonies for 7 days) in different seasons and at different sites by analyzing terminal restriction fragment length polymorphism (TRFLP) of 16S rRNA genes in bacterial community DNA. This method has been used successfully for the characterization of bacterial communities in marine samples [35, 60, 66] and has certain advantages over DGGE and 16S rRNA gene cloning in terms of its effectiveness, sensitivity, and consistency in differentiating microbial communities [9, 30, 60]. The variable and stable fractions of the bacteria communities were distinguished, and the possible identities of the stable bacterial associates were inferred using reference terminal restriction fragments (TRFs) derived from bacteria isolated from ecological relevant sites.
Materials and Methods
Sampling Site and Time
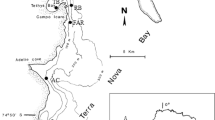
Bacterial communities were sampled from the surfaces of the sponge M. adhaerens and polystyrene petri dishes (as inanimate reference) at three sites in the eastern Hong Kong waters, namely, Three Fathoms Cove (site A, Fig. 1), Long Harbour (site B, Fig. 1), and Clear Water Bay (site C, Fig. 1), in April (spring), July (summer), October 2003 (autumn), and January 2004 (winter). The three sites are at least 11.7 km away from each other, and environmental variables are monthly monitored by the Environmental Protection Department of Hong Kong SAR government year around. Site A continuously receives discharges from the numerous water catchments in the area and has the lowest salinity (30.9 ppt), but has the highest temperature (24.7°C), nutrient contents (total nitrogen: 0.19 mg L–1, phosphorus: 0.03 mg L–1), pH (8.3), and chlorophyll a content (3.55 μg L–1) among the three sites. Site B is a relatively open area receiving prevailing easterly winds throughout the year with the lowest temperature (24.0°C), pH (8.1), dissolved oxygen (6.3 mg L–1), and total nitrogen (0.13 mg L–1) and phosphorus content (<0.02 mg L–1). Site C is a small enclosed bay area (opening of the bay is about 280 m) with relatively stagnant water, which has the highest salinity (32.3 ppt) but the lowest chlorophyll a content (1.66 μg L–1).

The location of sampling sites in the eastern Hong Kong waters. Site A: Three Fathoms Cove (22°26.91′N, 114°16.06′E); site B: Long Harbour (22°27.33′N, 114°21.00′E); site C: Clear Water Bay (22°15.92′N, 114°17.41′E).
Collection and Extraction of Bacterial Community DNA
During each sampling, four colonies of M. adhaerens, each with a surface area of ca. 40 cm2, were rinsed with autoclaved 0.22-μm-filtered seawater. The surfaces were swabbed with sterile cotton tips to collect bacterial community. Each cotton tip was immediately frozen in 0.8 mL of extraction buffer (100 mM of Tris–HCl, 100 mM of Na2–EDTA, 100 mM of Na2HPO4, 1.5 M of NaCl, 1% of cetyl trimethylammonium bromide; at pH 8) and transported to the laboratory. Reference bacterial communities were collected analogously from presterile polystyrene dishes, which had been deployed in the close vicinity (2 m) of the sponge colonies 7 days prior to the sampling. Bacterial cells in the samples were lysed in two freeze–thaw cycles using liquid nitrogen and a 65°C water bath. Total DNA was extracted and purified according to the sodium-dodecyl-sulfate-based method described in [99]. Purified DNA was dissolved in 50 μL of sterile H2O and kept at −20°C until use.
Polymerase Chain Reaction
The 16S rRNA genes in the DNA samples were amplified by polymerase chain reaction (PCR) using the primers 341F (5′-CCTACG GGAGGCAGCAG-3′) and 926R-Fam (5′-CCGTCAATT CCTTTRAGTTT-3′) [55]. The latter was labeled at the 5′ end with 6-carboxy fluorescein (Fam). Each PCR mixture contained 5 μL of DNA sample, 1.25 U of Taq polymerase (Amersham Biosciences, USA), 0.25 mM of dNTPs, 0.1 μM of each primer, 1.5 mM of MgCl2, and 2.5 μL of 10 × PCR buffer in a total volume of 25 μL. PCR was performed in the following thermal cycles: 95°C for 2 min (initial denaturation); 10 touchdown cycles of 95°C for 1 min (denaturation), 66°C (reduced to 57°C in increments of 1°C/cycle) for 1 min (annealing), and 72°C for 1 min (extension); additional 20 cycles with constant annealing temperature of 56°C; and 72°C for 5 min (final extension).
TRFLP Analysis of Bacterial Communities
Polymerase chain reaction products were cleaved with 10 U of the restriction enzyme MspI at 37°C for 6 h. Cleaved PCR products were kept at −20°C until purification using the Wizard® PCR preps DNA purification system (Promega, USA). Ten microliters of purified products together with 0.5-μL internal size standard (ET550-R, Amersham Biosciences) were denatured at 95°C for 2 min, snap cooled on ice, and subjected to electrophoresis on a MegaBACE genetic analyzer (Amersham) operated in the genotyping mode. After electrophoresis, the size of the fluorescently labeled TRFs was determined by comparison with the size standards (ET550-R, Amersham Biosciences) using the software Genetic Profiler (Amersham Biosciences). TRFs that were < 50, 100, or 250 fluorescence units in intensity, < 35 bp in size, and > 550 bp in size were excluded from statistical analysis to screen off background noise, to avoid pseudo-TRFs derived from primers, and to avoid inaccurate size determination, respectively [35, 60].
Estimation of Phylogenetic Affiliation of Bacteria in the Samples
To estimate the phylogenetic affiliation of the bacteria from which the TRFs were derived, the size of TRFs derived from the samples was compared to those derived from 20 bacterial strains that had been isolated from the surface of M. adhaerens [53]. Extraction of crude DNA from each individual bacterial strain followed [85], while the primers, restriction enzymes, and PCR conditions to obtain TRFs from each isolate were the same as mentioned above. In addition, an on-line analysis from the Ribosomal Database Project (RDP; http://wdcm.nig.ac.jp/RDP/html/analyses.html) [14] was employed to estimate the possible identity of the bacteria from which TRFs of interest were derived.
Statistical Analysis
The number of TRFs derived from the sponge and reference surfaces was compared by t-test. The similarity of TRF profiles among different samples was determined using nonmetric multidimensional scaling (MDS) and one-way analysis of similarities (ANOSIM) based on the size of the TRFs. Because PCR amplification methods may be biased toward numerically dominant bacteria or result in differential amplification that varies among taxa [81], the relative fluorescence intensities of TRFs may not be accurate measures of the relative abundances of population members within a given sample and were not used in the statistical analysis. Instead, similarity matrices based on the total number of TRFs observed in all samples and the presence or absence of these TRFs in individual samples were generated. The rank orders of similarity between different samples given in the similarity matrix were used to calculate a two-dimensional ordination. The degree of disagreement between the rank-order distances among bacterial communities as displayed by the MDS plot and the real distances as derived from the similarity matrix is defined by the stress value [13]. Furthermore, global differences among individual samples were tested by one-way ANOSIM that is a nonparametric analog to analysis of variance (ANOVA) without the assumption of normality and homogeneity [11]. Pairwise comparisons tested for differences between samples when ANOSIM was significant. The ANOSIM statistic R is calculated based on the difference of mean ranks between groups (r_B) and within groups (r_W) and lies between −1 and +1:
R values > 0.75 are generally interpreted as well separated, R > 0.5 as overlapping but clearly different, and R < 0.25 as barely separable at all [12]. Calculations were performed with the PRIMER v3.1 computer program (Plymouth Marine Laboratory, UK).
Results
TRF Profiles of Bacterial Communities
Figure 2 showed the representative electropherograms of TRFs derived from MspI digestion of PCR-amplified bacterial community DNA obtained from the sponge and reference surfaces. Under the combination of primers and restriction enzyme used in this study, the mean number of discernible TRFs derived from each type of samples ranged from 6 to 20 (Fig. 3). Generally speaking, numbers of TRFs derived from samples collected from site A (a total of 455 TRFs) and in winter (a total of 319 TRFs) were the highest among sites and seasons. For most of the cases, number of TRFs derived from the sponge and reference surfaces did not differ from each other significantly (Fig. 3).

Representative electropherograms of TRFs derived from MspI digestion of PCR-amplified bacterial community DNA obtained from the sponge (upper panel) and reference surfaces (lower panel). Samples for the sponge and reference surfaces were collected from site A during the first sampling in spring.

Number of TRFs derived from MspI digestion of PCR-amplified bacterial community DNA obtained from the sponge (open bars) and reference surfaces (closed bars) from different locations at different times. Data are expressed as the mean + 1 SD of four replicates. *Data that are significantly different from the sponge surface in t-test.
For temporal variation of bacterial community composition, MDS ordinations arranged the bacterial communities obtained from each season into two distinctive groups: one for the sponge surface and one for the reference surface (stress value = 0.11–0.14; Fig. 4a). The bacterial communities derived from the reference surface were further divided into three subgroups, each containing all replicate samples of the same site. The bacterial communities from the sponge surface showed no clear distinction among the individual sites (Fig. 4a). Similar grouping was obtained when the bacterial communities derived from each site were analyzed for seasonal variations; the sponge bacterial communities were all clearly separated from the reference bacterial communities (stress value = 0.15–0.18; Fig. 4b). Whereas the reference bacterial communities could be further grouped into separate seasons, the sponge bacterial communities showed no clear distinction among the seasons (Fig. 4b). The results of MDS ordinations were further supported by those of ANOSIM that detected significant differences between the sponge and reference samples as well as among the reference samples (R values = 0.60–1.00; Table 1) and that showed no clear distinction among the sponge samples from different sites and seasons (R values = 0.08–0.98; Table 1). Similar observation was found when TRF threshold intensity was set at 100 or 250 fluorescence units in statistical analysis (data not shown), and therefore, only the data set generated based on the threshold intensity at 50 fluorescence units was presented.


MDS ordinations of bacterial communities (4 replicates each) associated with the sponge surface (open symbols) and the reference surface (filled symbols) from (a) different sampling sites (Site A: triangles; Site B: squares; Site C: circles) at a particular sampling time, or form (b) different sampling times (Spring: rhombuses; Summer: crosses; Autumn: pentagons; Winter: hexagons) at a particular sampling site. The circles in dotted-line indicate sample groups of relatively high similarity.
Table 2 summarizes the occurrence of common TRFs among samples. The TRFs at 302 and 375 bp were found at all sampling sites and at all sampling times in both the sponge and reference samples. Some TRFs (e.g., 301, 370, 371, 422, and 423 bp) were found on both the sponge and reference samples from all sites for at least one sampling time (Table 2). The TRFs at 248, 310, and 311 bp were observed exclusively on the sponge surface at all sampling sites and at all sampling times (Table 2). No such exclusive distinctive TRFs were observed for the reference samples. Some other TRFs were associated with either the sponge (e.g., 150, 247, 249, 419, and 420 bp) or the reference samples (e.g., 300, 301, 303, 370, and 422 bp) at least at one sampling time (Table 2).
Phylogenetic Affiliation of Bacteria in the Samples
In total, 13 out of 20 bacteria isolated from the surface of the sponge in our previous study [53] generated TRFs that could be matched with the TRFs derived from the sponge bacterial community samples (Table 3). With the exception of S10 (Alteromonas alvinellae), which generated two TRFs, all other bacterial isolates generated single TRFs that ranged from 129 to 422 bp upon MspI digestion of PCR-amplified 16S rRNA gene (Table 3). The TRF at 302 bp in the sponge bacterial community samples could match with the TRFs of four isolates that belonged to the gram-positive division (S16, S17, and S18) and the α-subdivision of Proteobacteria (S15; Table 3), whereas the TRF at 314 bp could match with the TRFs of two isolates that belonged to the γ-subdivision of Proteobacteria (S5 and S20). The TRF at 301 bp could match with those of another two isolates that belonged to the α-subdivision of Proteobacteria (S7 and S8). Other TRFs at 150, 248, 310, 368, 371, and 422 bp could match with the TRFs of one isolate belonging to a variety of taxa including the γ-subdivision of Proteobacteria, the gram-positive group, and the Cytophaga-Flexibacter-Bacteroides group (Table 3). However, TRFs at 129 and 206 bp derived from the bacterial isolates were not observed in the TRFLP profiles of the sponge bacterial community samples.
Using the on-line analysis from the RDP [14], bacteria that could produce TRFs at particular size can be theoretically inferred. In most cases, one particular size of TRF could be generated from more than one sequence deposited in the database; for instance, about 200 sequences deposited in the database could theoretically generate TRF at 302 bp when digested with MspI. Sequences from all clones and some clinical strains were excluded from this study, and most of the strains listed in Table 4 came from environmental samples such as microbial mat, groundwater, deep-sea sediment, and marine- and freshwaters. After screening, 14 Enterococcus spp., 2 Abiotrophia spp., 2 Lactobacillus spp., and 3 other strains in the genera of Macrococcus, Marinbicaillus, and Tetragenococcus could generate TRF of 302 bp (Table 4). Similarly, strains from 23 different genera, e.g., Acidovorax, Burkholderia, and Herbaspirillum, would result in TRF at 310 bp, whereas 6 Brevundimonas spp. and 6 other strains in the genera of Caulobacter, Clostridium, Thiocystis, Marichromatium, Nevskia, and Paracoccus could produce TRF at 311 bp (Table 4). However, no bacterial sequence deposited in the database could generate TRFs at 248 and 375 bp (Table 4).
Discussion
Statistical analysis of TRF profiles revealed significant differences in genotype diversity of the bacterial communities between the reference surfaces and sponge surfaces at all sites and seasons (Fig. 4; reference surface vs sponge surface, Table 1). The diversity of the bacterial communities on the reference surface showed clear seasonality and site specificity, which were indicated by the segregation of the replicate bacterial communities derived from different sites and seasons in MDS ordinations (Fig. 4), and further supported by the significantly high R values in ANOSIM (sponge surface vs reference surface, Table 1). These site and seasonal differences of bacterial community structure could be a result of the changes in surface-related attributes, prevailing environmental conditions, and/or the availability of bacterial colonizers in the water column. One may question if the materials (made of polystyrene that is a rather inert, hydrophobic material) used for the reference surface in this study could create arbitrary results. Based on our previous observations and the works in [88], the hydrophobicity/hydrophilicity of a surface may influence the composition of bacterial community at the initial stage of colonization, its effect often diminishes rapidly after the initial colonization, and the subsequent colonization by other bacteria is mainly influenced by environmental and biological factors. Therefore, we strongly believe that the observed seasonality and site specificity was a result of the differences of environmental conditions prevailing at each sampling site and time, including seawater temperature, the amount of terrestrial runoff, flow speed, turbidity, water chemistry, and nutrient content. All these factors, either singly or in combinations, can affect the composition of indigenous bacterial populations in the water column and thus the composition of bacterial communities that would eventually colonize the reference surface. It would be equally interesting to investigate the spatial and temporal variations of bacterial communities in the water columns to have a better understanding on how these variations influence the bacterial communities developed on the reference as well as on the sponge surfaces.
In contrast to the obvious seasonality and site specificity of the bacterial community on the reference surfaces, the bacterial communities on the sponge surfaces at individual sites or seasons were generally less distinguishable among different sites or seasons as shown by the interspersed MDS ordinations of the replicate bacterial communities in Fig. 4 and by the relatively low R values in ANOSIM (sponge surface vs sponge surface, Table 1). In contrast to the association with the inert reference surface, the bacterial community on the sponge surface may be influenced more by complex surface-related (i.e., sponge-related) attributes than that of environmental factors. These attributes may include the continuous release of bioactive metabolites (as chemical defense against bacterial epibiosis and/or the enrichment of specific bacterial types), the texture and topography of the sponge surface, and the presence of communistic meiofauna. The quantitative and qualitative differences in these attributes among individual sponges may explain the generally high variability of the bacterial community profiles derived from the sponge surface.
Although the bacterial communities on the sponge surfaces showed a certain degree of variability, eight TRFs in common were found among the three sites and were only found in the sponge-surface-associated bacterial communities (Table 2). Five of these TRFs (at 150, 247, 249, 419, and 420 bp) were found only in one or few sampling occasions; the sporadic occurrence of these TRFs suggests an occasional or less consistent association between the bacteria producing these TRFs and the sponge surface. On the other hand, the other three TRFs (at 248, 310, and 311 bp) were present consistently on the sponges at all sites and all seasons. The consistent association between the bacteria producing these three TRFs and the sponge surface may be the result of the selective forces (such as sponge's secondary metabolites) derived from the sponge or of the specific adaptation of these bacteria to the microenvironment on the sponge surface. In fact, many sponges may eliminate or nurture specific bacterial groups by the production of secondary metabolites [3, 10, 36, 37, 59]. When we compared TRFs of specific size with TRFs derived from the pure bacterial strains (Table 3) isolated from the surface of M. adhaerens [53], we deduced that the highly consistent TRFs at 248 and 310 bp may belong to Alteromonas alvinellae (S6). Crosby and Criddle [19] also pointed out that multiple TRFs could be derived from the same species because of ribosomal operon heterogeneity. On the other hand, the RDP on-line analysis indicated that the highly consistent TRFs at both 310 and 311 bp may also be derived from a variety of bacteria, but most of these bacteria were in the same genus. This may be caused by the possibility that closely related bacteria may share similar DNA sequence and have same restriction enzyme cutting site, thus generating TRFs of same length upon digestion by one restriction enzyme. The resolution of TRFLP analysis in differentiating different bacteria can be improved by multiple digests using several restriction enzymes individually followed by the use of TRFLP phylogenetic assignment tool [22, 50]. Although our data here cannot identify the bacterial species that are consistently associated with the sponge surface, our results suggested that the highly consistent and specific bacterial TRFs observed on the sponge surface might be derived from a specific group of closely related bacteria. The highly consistent association of these bacteria with the sponge surface might reflect a beneficial interrelationship that enhances the survival of either one or both parties.
Two TRFs (at 302 and 375 bp) consistently appeared on both the sponges and reference surfaces at all sites and all seasons (Table 2). They were also found in the bacterial communities of the water columns of the three sites (data not shown). These TRFs may represent certain indigenous bacterial colonizers available in the water column throughout the year and colonized equally well on both the sponge and reference surfaces. Comparison of the TRFs to those derived from the bacteria isolated from ecologically relevant sites and to those estimated by the on-line analysis revealed that TRF at 302 bp might be derived from a number of bacteria in the gram-positive group (Tables 3 and 4). The inability of matching TRF at 375 bp to any bacterial isolates or sequences in the database might suggest that the bacteria deriving this particular size of TRF may not be culturable. It is also possible that small size discrepancies resulted from fragment sizing in analysis lead to the inability in matching. On the other hand, five other TRFs (at 301, 370, 371, 422, and 423 bp) that appeared on both surfaces only at particular seasons suggested a seasonal variation in the composition of indigenous bacterial colonizers.
Five TRFs (at 300, 301, 303, 370, and 422 bp) were exclusively associated with the reference surfaces at least at one sampling time (Table 2). However, unlikely the consistent association of some TRFs with the sponge surface, none of these five TRFs was present on the reference surface year round, suggesting possible fluctuations in bacterial communities with environmental factors.
The reference surface bacterial communities were developed on polystyrene dishes deployed in the close vicinity of the sponge for 7 days. One may also argue that the bacterial community developed under this condition may not be comparable to that on the sponge that has developed over months or years. This was not the case for our study for the following reasons. Before we decided to use 7 days as the submersion duration, we had already carried out a preliminary experiment, in which we had provided a “clean” sponge surface by removing its surface-associated bacteria and then resubmerged it for 7 days to allow the development of microbial film. We found that TRF patterns of the bacterial communities before and after cleaning and resubmersion were highly similar (similarity ∼80%, Fig. 5), indicating that the development of bacterial film on the sponge surface could reach a “stable” stage after 7 days. Similarly, Qian et al. [66] showed that bacterial communities developed on polystyrene dishes for 6, 9, and 12 days were highly similar, suggesting that a stable bacterial community could be obtained after 6 days of submersion in Hong Kong waters. Furthermore, it is almost impossible to obtain a “pure” microbial film after a longer time of submersion because other organisms such as invertebrate larvae will colonize the filmed surface. These organisms will carry their own bacteria added to the reference bacterial community and complicate the analysis of bacterial community originated from the reference dish only.

Representative electropherograms of TRFs derived from MspI digestion of PCR-amplified bacterial community DNA obtained from the sponge surfaces. Upper and lower panels show the bacterial communities before and after resubmersion, respectively, in a preliminary experiment.
In summary, high degree of site and seasonal specificities in the reference bacterial communities demonstrated in this study might be attributed to the differences in environmental conditions. In contrast, the sponge-surface-associated bacterial communities were less distinguishable from each other but clearly differed from those on the reference ones, suggesting a rather distinctive and stable bacteria–sponge association irrespective of the environmental conditions. The consistent association of unique bacterial types with M. adhaerens over three geographically separate sites at four seasons provides direct evidence of a highly stable association between the two parties. Future works on 16S rDNA clone library of the surface-associated bacterial community will help us identify the unique bacterial associates of the sponge.
Reference
Abraham, WR, Strompl, C, Meyer, H, Lindholst, S, Moore, ER, Christ, R, Vancanneyt, M, Tindall, BJ, Bennasar, A, Smit, J, Tesar, M (1999) Phylogeny and polyphasic taxonomy of Caulobacter species. Proposal of Maricaulis gen. nov. with Maricaulis maris (Poindexter) comb. nov. as the type species, and emended description of the genera Brevundimonas and Caulobacter. Int J Syst Bacteriol 49: 1053–1073
Achouak, W, Christen, R, Barakat, M, Martel, MH, Heulin, T (1999) Burkholderia caribensis sp. nov., an exopolysaccharide-producing bacterium isolated from vertisol microaggregates in Martinique. Int J Syst Bacteriol 49: 787–794
Amade, P, Charroin, C, Baby, C, Vacelet, J (1987) Antimicrobial activities of marine sponges from the Mediterranean Sea. Mar Biol 94: 271–275
Anzai, Y, Kim, H, Park, JY, Wakabayashi, H, Oyaizu, H (2000) Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int J Syst Evol Microbiol 50(4): 1563–1589
Back, W, Bohak, I, Ehrmann, M, Ludwig, W, Schleifer, KH (1996) Revival of the species Lactobacillus lindneri and the design of a species specific oligonucleotide probe. Syst Appl Microbiol 19: 322–325
Bauernfeind, A, Schneider, I, Jungwirth, R, Roller, C (1999) Discrimination of Burkholderia multivorans and Burkholderia vietnamiensis from Burkholderia cepacia genomovars I, III, and IV by PCR. J Clin Microbiol 37(5): 1335–1339
Borowitzka, MA, Hinde, R, Prionet, F (1988) Carbon fixation by the sponge Dysidea herbacea and its endosymbiont Oscillatoria spongeliae. In: Choat, JH et al. (Eds.) Proc 6th Int Coral Reef Symposium, Townsville, Australia, pp 151–155
Brett, PJ, Deshazer, D, Woods, DE (1997) Characterization of Burkholderia pseudomallei and Burkholderia pseudomallei-like strains. Epidemiol Infect 118(2): 37–148
Brodie, E, Edwards, S, Clipson, N (2002) Bacterial community dynamics across a floristic gradient in a temperate upland grassland ecosystem. Microb Ecol 44: 260–270
Burkholder, PR, Rützler, K (1969) Antimicrobial activity of some marine sponges. Nature 222: 983–984
Clarke, KR (1993) Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 18: 117–143
Clarke, KR, Gorley, RN (2001) PRIMER v5: User Manual/Tutorial. PRIMER-E Ltd., Plymouth, UK
Clarke, KR, Warwick, RM (1994) Changes in Marine Communities: An Approach to Statistical Analysis and Interpretation. Bourne Press, Bournemouth
Cole, JR, Chai, B, Farris, RJ, Wang, Q, Kulam, SA, McGarrell, DM, Garrity, GM, Tiedje, JM (2005) The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res 33(Database issue): D294–D296
Collins, MD, Jones, D, Farrow, JAE, Kilpper-Balz, R, Schleifer, KH (1984) Enterococcus avium nom. rev., comb. nov.; E. casseliflavus nom. rev., comb. nov.; E. durans nom. rev., comb. nov.; E. gallinarum comb. nov.; and E. malodoratus sp. nov. Int J Syst Bacteriol 34: 220–223
Collins, MD, Farrow, JAE, Jones, D (1986) Enterococcus mundtii sp. nov. Int J Syst Bacteriol 36: 8–12
Collins, MD, Facklam, RR, Farrow, JA, Williamson, R (1989) Enterococcus raffinosus sp. nov., Enterococcus solitarius sp. nov. and Enterococcus pseudoavium sp. nov. FEMS Microbiol Lett 48: 283–288
Collins, MD, Rodrigues, UM, Pigott, NE, Facklam, RR (1991) Enterococcus dispar sp. nov. a new Enterococcus species from human sources. Lett Appl Microbiol 12: 95–98
Crosby, LD, Criddle, CS (2003) Understanding bias in microbial community analysis techniques due to rrn operon copy number heterogeneity. BioTechniques 34: 790–797
Dekhil, B, Cahill, M, Stackebrandt, E, Sly, LI (1997) Transfer of Conglomeromonas largomobilis subsp. largomobilis to the genus Azospirillum as Azospirillum largomobile comb. nov., and elevation of Conglomeromonas largomobilis subsp. parooensis to the new type species of Conglomeromonas, Conglomeromonas parooensis sp. nov. Syst Appl Microbiol 20: 72–77
Devriese, LA, Dutta, GN, Farrow, JAE, Van De Kerckhove, A, Phillips, BA (1983) Streptococcus cecorum, a new species isolated from chickens. Int J Syst Bacteriol 33: 772–776
Dunbar, J, Ticknor, LO, Kuske, CR (2000) Assessment of microbial diversity in four southwestern United Stated soils by 16s rRNA gene terminal restriction fragment analysis. Appl Environ Microbiol 66: 2943–2950
Engel, S, Pawlik, JR (2000) Allelopathic activities of sponge extracts. Mar Ecol Prog Ser 207: 273–281
Ennahar, SE, Cai, Y (2005) Biochemical and genetic evidence for the transfer of Enterococcus solitarius Collins et al. 1989 to the genus Tetragenococcus as Tetragenococcus solitarius comb. nov. Int J Syst Evol Microbiol 55: 589–592
Evvyernie, D, Yamazaki, S, Morimoto, K, Karita, S, Kimura, T, Sakka, K, Ohmiya, K (2000) Identification and characterization of Clostridium paraputrificum M-21, a chitinolytic, mesophilic and hydrogen-producing bacterium. J Biosci Bioeng 89: 596–601
Farrow, JAE, Collins, MD (1985) Enterococcus hirae, a new species that includes amino acid assay strain NCDO 1258 and strains causing growth depression in young chickens. Int J Syst Bacteriol 35: 73–75
Friedrich, AB, Merkert, H, Fendert, T, Hacker, J, Proksch, P, Hentschel, U (1999) Microbial diversity in the marine sponge Aplysina cavernicola (formerly Verongia cavernicola) analyzed by fluorescence in situ hybridization (FISH). Mar Biol 134: 461–470
Friedrich, AB, Fischer, I, Proksch, P, Hacker, J, Hentschel, U (2001) Temporal variation of the microbial community associated with the Mediterranean sponge Aplysina aerophoba. FEMS Microbiol Ecol 38: 105–113
Fuerst, JA, Webb, RI, Garson, MJ, Hardy, L, Reiswig, HM (1999) Membrane-bounded nuclear bodies in a diverse range of microbial symbionts of Great Barrier Reef sponges. Mem Queensl Mus 44: 193–203
Fuhrman, JA, Griffith, JF, Schwalbach, MC (2002) Prokaryotic and viral diversity pattern in marine plankton. Ecol Res 17: 183–194
Gardan, L, Dauga, C, Prior, P, Gillis, M, Saddler, GS (2000) Acidovorax anthurii sp. nov., a new phytopathogenic bacterium which causes bacterial leaf-spot of anthurium. Int J Syst Evol Microbiol 50: 235–246
Garson, MG, Flowers, AE, Webb, RI, Charan, RD, McCaffrey, EJ (1998) A sponge/dinoflagellate association in the haposclerid sponge Haliclona sp.: cellular origin of cytotoxic alkaloids by percoll density gradient fractionation. Cell Tissue Res 293: 365–373
Glockner, FO, Babenzien, HD, Amann, R (1998) Phylogeny and identification in situ of Nevskia ramosa. Appl Environ Microbiol 64: 1895–1901
Gilbert, JJ, Allen, HL (1973) Chlorophyll and primary productivity of some green, freshwater sponges. Int Rev Gesamten Hydrobiol 58(5): 633–658
Harder, T, Lau, SCK, Dobretsov, S, Fang, TK, Qian, PY (2003) A distinctive epibiotic bacterial community on the soft coral Dendronephthya sp. and antibacterial activity of coral tissue extracts suggest chemical mechanism against bacterial epibiosis. FEMS Microbiol Ecol 43: 337–347
Harder, T, Lau, SCK, Tam, WY, Qian, PY (2004) A bacterial culture-independent method to investigate chemically mediated control of bacterial epibiosis in marine invertebrates by using TRFLP analysis and natural bacterial populations. FEMS Microbiol Ecol 47: 93–99
Hentschel, U, Schmid, M, Wagner, M, Fieseler, L, Gernert, C, Hacker, J (2001) Isolation and phylogenetic analysis of bacteria with antimicrobial activities from the Mediterranean sponges Aplysina aerophoba and Aplysina cavernicola. FEMS Microbiol Ecol 35: 305–312
Hentschel, U, Hopke, J, Horn, M, Friederich, AB, Wagner, M, Hacker, J, Moore, BS (2002) Molecular evidence for a uniform microbial community in sponges from different oceans. Appl Environ Microbiol 68: 4431–4440
Hentschel, U, Fieseler, L, Wehrl, M, Gernert, C, Steinert, M, Hacker, J, Horn, M (2003) Microbial diversity of marine sponges. In: Müller WEG (Ed.) Marine Molecular Biotechnology, Springer-Verlag, Berlin, pp 59–88
Hippe, H, Hagelstein, A, Kramer, I, Swiderski, J, Stackebrandt, E (1999) Phylogenetic analysis of Formivibrio citricus, Propionivibrio dicarboxylicus, Anaerobiospirillum thomasii, Succinimonas amylolytica and Succinivibrio dextrinosolvens and proposal of Succinivibrionaceae fam. nov. Int J Syst Bacteriol 49: 779–782
Imamura, N, Nishijima, M, Adachi, K, Sano, H (1993) Novel antimycin antibiotics, urauchimycins A and B, produced by marine actinomycete. J Antibiot (Tokyo) 46: 241–246
Imhoff, JF, Suling, J, Petri, R (1998) Phylogenetic relationships among the Chromatiaceae, their taxonomic reclassification and description of the new genera Allochromatium, Halochromatium, Isochromatium, Marichromatium, Thiococcus, Thiohalocapsa and Thermochromatium. Int J Syst Bacteriol 48: 1129–1143
Imhoff, JF, Stöhr, R (2003) Sponge-associated bacteria: general overview and special aspects of bacteria associated with Halichondria panacea. In: Müller WEG (Ed.) Sponges (Porifera), Springer-Verlag, Berlin, pp 35–57
Jayatilake, GS, Thornton, MP, Leonard, AC, Grimwade, JE, Baker, BJ (1996) Metabolites form an Antarctic sponge-associated bacterium Pseudomonas aeruginosa. J Nat Prod 59: 293–296
Jiao, Z, Kawamura, Y, Mishima, N, Yang, R, Li, N, Liu, X, Ezaki, T (2003) Need to differentiate lethal toxin-producing strains of Burkholderia gladioli, which cause severe food poisoning: description of B. gladioli pathovar cocovenenans and an emended description of B. gladioli. Microbiol Immunol 47(12): 915–925
Kalmbach, S, Manz, W, Wecke, J, Szewzyk, U (1999) Aquabacterium gen. nov., with description of Aquabacterium citratiphilum sp. nov., Aquabacterium parvum sp. nov. and Aquabacterium commune sp. nov., three in situ dominant bacterial species from the Berlin drinking water system. Int J Syst Bacteriol 49: 769–777
Kanamoto, T, Sato, S, Inoue, M (2000) Genetic heterogeneities and phenotypic characteristics of strains of the genus Abiotrophia and proposal of Abiotrophia para-adiacens sp. nov. J Clin Microbiol 38(2): 492–498
Kandler, O, Kunath, P (1983) Lactobacillus kefir sp. nov., a component of the microflora of kefir. Syst Appl Microbiol 4: 286–294
Kelly, SR, Jensen, PR, Henkel, TP, Fenical, W, Pawlik, JR (2003) Effects of Caribbean sponge extracts on bacterial attachment. Aquat Microb Ecol 31(2): 175–182
Kent, AD, Smith, DJ, Benson, BJ, Triplett, EW (2003) Web-based phylogenetic assignment tool for analysis of terminal restriction fragment length polymorphism profiles of microbial communities. Appl Environ Microbiol 69: 6768–6776
Kloos, WE, Ballard, DN, George, CG, Webster, JA, Hubner, RJ, Ludwig, W, Schleifer, KH, Fiedler, F, Schubert, K (1998) Delimiting the genus Staphylococcus through description of Macrococcus caseolyticus gen. nov., comb. nov. and Macrococcus equipercicus sp. nov., and Macrococcus bovicus sp. nov. and Macrococcus carouselicus sp. nov. Int J Syst Bacteriol 48: 859–877
Kubanek, J, Whalen, KE, Engel, S, Kelly, SR, Henkel, TP, Fenical, W, Pawlik, JR (2002) Multiple defensive roles for triterpene glycosides from two Caribbean sponges. Oecologia 131: 125–136
Lee, OO, Qian, PY (2003) Chemical control of bacterial epibiosis and larval settlement of Hydrodies elegans in the red sponge Mycale adhaerens. Biofouling 19(Supplement): 171–180
Lee, OO, Qian, PY (2004) Potential control of bacterial epibiosis on the surface of the sponge Mycale adhaerens. Aquat Microb Ecol 34: 11–21
Liu, WT, Marsh, TL, Cheng, K, Forney, LJ (1997) Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl Environ Microbiol 63: 4516–4522
Madigan, MT, Jung, DO, Woese, CR, Achenbach, LA (2000) Rhodoferax antarcticus sp. nov., a moderately psychrophilic purple nonsulfur bacterium isolated from an Antarctic microbial mat. Arch Microbiol 173: 269–277
Margot, HM, Acebal, C, Toril, E, Amils, R, Fernandez, Puentes, JL (2002) Consistent association of crenarchaeal Archaea with sponge of the genus Axinella. Mar Biol 140: 739–745
Martinez-Murcia, AJ, Collins, MD (1991) Enterococcus sulfureus, a new yellow-pigmented Enterococcus species. FEMS Microbiol Lett 80: 69–74
McCaffrey, EJ, Endean, R (1985) Antimicrobial activity of tropical and subtropical sponges. Mar Biol 89: 1–8
Moesender, MM, Arrieta, JM, Muyzer, G, Winter, C, Herndl, GJ (1999) Optimization of terminal-restriction fragment length polymorphism analysis for complex marine bacterioplankton communities and comparison with denaturing gradient gel electrophoresis. Appl Environ Microbiol 65: 3518–3525
Park, JK, Shimono, K, Ochiai, N, Shigeru, K, Kurita, M, Ohta, Y, Tanaka, K, Matsuda, H, Kawamukai, M (1999) Purification, characterization, and gene analysis of a chitosanase (ChoA) from Matsuebacter chitosanotabidus 3001. J Bacteriol 181: 6642–6649
Pfennig, N, Trper, HG (1971) Type and neotype strains of the species of phototrophic bacteria maintained in pure culture. Int J Syst Bacteriol 21: 19–24
Pompei, R, Berlutti, F, Thaller, MC, Ingianni, A, Cortis, G, Dainelli, B (1992) Enterococcus flavescens sp. nov., a new species of enterococci of clinical origin. Int J Syst Bacteriol 42: 365–369
Poindexter, JS (1964) Biological properties and classification of the Caulobacter group. Bacteriol Rev 28: 231–295
Preston, CM, Wu, KY, Molinski, TF, DeLong, EF (1996) A psychrophilic crenarchaeon inhabits a marine sponge: Cenarchaeum symbiosum gen. nov., sp. nov. Proc Natl Acad Sci USA 93: 6241–6246
Qian, PY, Thiyagarajan, V, Lau, SCK, Cheung, SCK (2003) Relationship between bacterial community profile in biofilm and attachment of the acorn barnacle Balanus amphitrite. Aquat Microb Ecol 33: 225–237
Rainey, FA, Kelly, DP, Stackebrandt, E, Burghardt, J, Hiraishi, A, Katayama, Y, Wood, AP (1999) A re-evaluation of the taxonomy of Paracoccus denitrificans and a proposal for the combination Paracoccus pantotrophus comb. nov. Int J Syst Bacteriol 49: 645–651
Rodrigues, U, Collins, MD (1990) Phylogenetic analysis of Streptococcus saccharolyticus based on 16S rRNA sequencing. FEMS Microbiol Lett 59: 231–234
Ruger, HJ, Fritze, D, Sproer, C (2000) New psychrophilic and psychrotolerant Bacillus marinus strains from tropical and polar deep-sea sediments and emended description of the species. Int J Syst Evol Microbiol 50: 1305–1313
Santavy, DL (1985) The symbiotic relationship between a blue-pigmented bacterium and the coral reef sponge, Terpiox granulose. In: Harmelin Vivien, M, Salvat, B (Eds.) Proc Fifth Int Coral Reef Congress, Tahiti, vol 5. Antenne Museum Ephe, Moorea, Tahiti, pp 135–140
Santavy, DL, Willenz, P, Colwell, RR (1990) Phenotypic study of bacteria associated with the Caribbean sclerosponge, Ceratoporella nicholsoni. Appl Environ Microbiol 56: 1750–1762
Sarà, M, Liaci, L (1964) Symbiotic association between zooxanthellae and two marine sponges of the genus Cliona. Nature 203: 321
Sato, S, Kanamoto, T, Inoue, M (1999) Abiotrophia elegans strains comprise 8% of the nutritionally variant streptococci isolated from the human mouth. J Clin Microbiol 37(8): 2553–2556
Schleifer, KH, Kilpper-Balz, R (1984) Transfer of Streptococcus faecalis and Streptococcus faecium to the genus Enterococcus nom. rev. as Enterococcus faecalis comb. nov. and Enterococcus faecium comb. nov. Int J Syst Bacteriol 34: 31–34
Schleifer, KH, Schuler, D, Spring, S, Weizenegger, M, Amann, R, Ludwig, W, Kohler, M (1991) The genus Magnetospirillum gen. nov. description of Magnetospirillum gryphiswaldense sp. nov. and transfer of Aquaspirillum magnetotacticum to Magnetospirillum magnetotacticum comb. nov. Syst Appl Microbiol 14: 379–385
Schulze, R, Spring, S, Amann, R, Huber, I, Ludwig, W, Schleifer, KH, Kampfer, P (1999) Genotypic diversity of Acidovorax strains isolated from activated sludge and description of Acidovorax defluvii sp. nov. Syst Appl Microbiol 22: 205–214
Segers, P, Vancanneyt, M, Pot, B, Torck, U, Hoste, B, Dewettinck, D, Falsen, E, Kersters, K, De Vos, P (1994) Classification of Pseudomonas diminuta Leifson and Hugh 1954 and Pseudomonas vesicularis Busing, Doll, and Freytag 1953 in Brevundimonas gen. nov. as Brevundimonas diminuta comb. nov. and Brevundimonas vesicularis comb. nov., respectively. Int J Syst Bacteriol 44: 499–510
Stahl, DA, Key, R, Flesher, B, Smit, J (1992) The phylogeny of marine and freshwater caulobacters reflects their habitat. J Bacteriol 174: 2193–2198
Stierle, DB, Stierle, AA (1992) Pseudomonic acid derivatives from a marine bacterium. Experientia 44: 1165–1169
Suyama, T, Shigematsu, T, Takaichi, S, Nodasaka, Y, Fujikawa, S, Hosoya, H, Tokiwa, Y, Kanagawa, T, Hanada, S (1999) Roseateles depolymerans gen. nov., sp. nov., a new bacteriochlorophyll a-containing obligate aerobe belonging to the beta-subclass of the Proteobacteria. Int J Syst Bacteriol 49: 449–457
Suzuki, MT, Giovanni, SJ (1996) Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62: 625–630
Thacker, RW, Starnes, S (2003) Host specificity of the symbiotic cyanobacterium Oscillatoria spongeliae in marine sponge, Dysides spp. Mar Biol 142: 643–648
Thoms, C, Horn, M, Wagner, M, Hentschel, U, Proksch, P (2003) Monitoring microbial diversity and natural products profiles of the sponge Aplysina cavernicola following transplantation. Mar Biol 42: 685–692
Vacelet, J, Donadey, C (1977) Electro microscope study of the association between some sponges and bacteria. J Exp Mar Biol Ecol 30: 301–314
Valsecchi, E (1998) Tissue boiling: a short-cut in DNA extraction for large-scale population screenings. Mol Ecol 7: 1243–1245
van der Meer, JR, Werlen, C, Nishino, SF, Spain, JC (1998) Evolution of a pathway for chlorobenzene metabolism leads to natural attenuation in contaminated groundwater. Appl Environ Microbiol 64(11): 4185–4193
Viallard, V, Poirier, I, Cournoyer, B, Haurat, J, Wiebkin, S, Ophel-Keller, K, Balandreau, J (1998) Burkholderia graminis sp. nov., a rhizospheric Burkholderia species, and reassessment of Pseudomonas phenazinium, Pseudomonas pyrrocinia and Pseudomonas glathei as Burkholderia. Int J Syst Bacteriol 48: 549–563
Wahl, M (1989) Marine epibiosis. I. Fouling and antifouling: some basic aspects. Mar Ecol Prog Ser 58: 175–189
Webster, NS, Hill, RT (2001) The culturable microbial community of the Great Barrier Reef sponge Rhopaloeides odorabile is dominated by α-proteobacterium. Mar Biol 138: 843–851
Wilkinson, CR (1978) Microbial associations in sponges. II. Numerical analysis of sponge and water bacterial populations. Mar Biol 49: 169–176
Wilkinson, CR, Garrone, R (1980) Nutrition in marine sponges. Involvement of symbiotic bacteria in the uptake of dissolved carbon. In: Smith, DC, Tiffon, Y (Eds.) Nutrition in the Lower Metazoa. Pergamon Press, Oxford, pp 157–161
Wilkinson, CR, Nowak, M, Austin, B, Colwell, RR (1981) Specificity of bacterial symbionts in Mediterranean and Great Barrier Reef sponges. Microb Ecol 7: 13–21
Willems, A, De Ley, J, Gillis, M, Kersters, K (1991) Comamonadaceae, a new family encompassing the acidovorans rRNA complex, including Variovorax paradoxus gen. nov., comb. nov., for Alcaligenes paradoxus (Davis 1969). Int J Syst Bacteriol 41: 445–450
Willems, A, Gillis, M, De Ley, J (1991) Transfer of Rhodocyclus gelatinosus to Rubrivivax gelatinosus gen. nov., comb. nov., and phylogenetic relationships with Leptothrix, Sphaerotilus natans, Pseudomonas saccharophila, and Alcaligenes latus. Int J Syst Bacteriol 41: 65–73
Williamson, CE (1979) An ultrastructural investigation of algal symbiosis in white and green Spongilla lacustris (L.) (Porifera: Spongillidae). Trans Am Microsc Soc 98(1): 59–77
Wilson, DM, Puyana, M, Fenical, W, Pawlik, JR (1999) Chemical defense of the Caribbean reef sponge Axinella corrugata against predatory fishes. J Chem Ecol 25(12): 2811–2824
Xie, CH, Yokota, A (2005) Reclassification of Alcaligenes latus IAM 12599T, IAM 12664, and Pseudomonas saccharophila as Azohydromonas lata gen. nov. comb. nov., Azohydromonas australica sp. nov., and Pelomonas saccharophila gen. nov. comb. nov. Int J Syst Evol Microbiol 55: 2419–2425
Zhang, H, Hanada, S, Shigematsu, T, Shibuya, K, Kamagata, Y, Kanagawa, T, Kurane, R (2000) Burkholderia kururiensis sp. nov., a trichloroethylene (TCE)-degrading bacterium isolated from an aquifer polluted with TCE. Int J Syst Evol Microbiol 50: 743–749
Zhou, J, Bruns, MA, Tiedje, JM (1996) DNA recovery from soils of diverse composition. Appl Environ Microbiol 62: 316–322
Acknowledgments
The authors thank the Environmental Protection Department of Hong Kong for providing data on water quality in the study areas. They also thank V. Thiyagarajan for consultation of statistical analysis and Y. K. Tam for assistance in TRFLP analysis. The study was supported by RGC grant (HKUST6240/04M) to P. Y. Qian.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee, O.O., Lau, S.C.K. & Qian, PY. Consistent Bacterial Community Structure Associated with the Surface of the Sponge Mycale adhaerens Bowerbank. Microb Ecol 52, 693–707 (2006). https://doi.org/10.1007/s00248-006-9077-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-006-9077-1