
Abstract
Microbial communities in the sediment and associated with the dominant type of standing dead plant were collected from the high marsh zones of 10 sites along the eastern coast of the United States from Maine to Florida. Microbial community composition was examined using T-RFLP, and bacterial and fungal abundance was determined microscopically. Within the sediment, community composition was strongly correlated with latitude, indicating that biogeographical factors are important determinants of sediment community composition, whereas abundance was positively and strongly correlated with sediment organic matter content. A strong biogeographical effect was observed for both bacterial and fungal abundance on standing dead plants, but there was no clear relationship between community composition and latitude. Microbial community composition was more similar among plants of the same type (i.e., related plant species) suggesting that plant type (i.e., substrate quality) is primarily responsible for the determining community composition on standing dead plants.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The earth is inhabited by an incredibly diverse assemblage of microorganisms that govern the rates and extents of all the biogeochemical cycles important for the existence of higher life. The prokaryotic biomass is estimated to be as much as half of the total biomass of the earth [71]. Estimates of the numbers of different types of microbes approach 2 × 106 for the oceans, and 4 × 106 per ton for soils [15]. Although Beijerink’s dictum that “everything is everywhere; the milieu selects” [3, 54] is still as true as at the time it was presented, the statement foreshadowed, but underestimated, the breadth of variability in both phylogeny and function that we are currently coming to realize. Bacteria may be found everywhere, but they are not uniformly distributed in either space or time. Large numbers of heretofore-undescribed genomes have been discovered using ribosomal DNA sequences as the criteria for identity, although the number of sequences actually recovered thus far is small [28] compared with the estimated total that likely exist [15]. The number of ecosystem functions that microbes perform is large, but the numbers of functions cannot match the number of types of organisms estimated to inhabit the earth [58]. Obviously, different types of organisms must often share similar functions. If the niche concept holds true for microbes (and there is no evidence that it does not), it is easily conceivable that the community selected will not necessarily be the same from place to place even though the environmental conditions are the same. It is clear that on a gross scale, microbial communities from different environments are indeed different: oceanic samples contain a very different suite of organisms from those recovered from agricultural soils [12, 15, 24]. Yet, it is equally conceivable that communities within similar habitat types from spatially distinct locations will be more similar to one another than communities from different habitats that are close together. Thus, variation in community composition within a habitat would be less than variation across habitat types of the same area (e.g., [21]). These ideas lead to the hypothesis that within a habitat type, microbial community composition may exhibit biogeography.
We report here the results of an evaluation of the microbial communities in two salt marsh habitat types, standing-dead plants and sediments in the high marsh zones of ten sites from Maine to Florida on the east coast of the United States. The standing-dead materials at these sites are part of a broader study to characterize the development of microbial decay communities within similar habitats and among different habitats to determine whether microbial communities exhibit biogeography. In part because of their variation in latitude, the sites used in this study are characterized by steep gradients in temperature, precipitation frequency and amount, humidity, salinity of sediment and inundating water, and light intensity.
The activities of microbial communities respond to a wide variety of biotic and abiotic factors that include substrate quality (e.g., [25, 57, 62, 65, 67]), substrate particle size [64], edaphic conditions [29, 67], and climate [8, 9, 33, 45]. It is reasonable to hypothesize that these same factors influence not only activity but also community composition. For example, a recent study demonstrated the importance of soil type, itself the product of biotic and abiotic influences, as a major factor in the composition and activity of bacterial communities in arable soils [24]. If similar microbial communities develop on similar marsh plant species or plant types (e.g., grasses vs reeds) across the various sites, substrate quality is implicated as the primary determinant of community composition on standing-dead plants. Conversely, if the microbial communities that develop on standing-dead biomass of the same plant species are compositionally different across sites, then environmental factors (e.g., temperature, precipitation, salinity) would be presumed to be the principal determinants of community composition. If microbial communities of similar composition are detected in the sediment but not on the standing-dead biomass, then environmental factors may constrain the type of microbial communities responsible for litter decomposition. Even though microbial activity and community composition may vary across sites for the same plant type, the decay processes may be very similar because of degeneracy within microbiological systems [17].
Materials and Methods
Site Descriptions
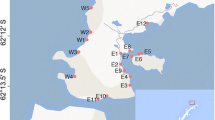
Ten sites participated in this comparative study (Fig. 1). The sites included all four active Atlantic coastal Long Term Ecological Research sites (Plum Island Estuary, Virginia Coast Reserve, Georgia Coastal Ecosystem, and Florida Coastal Everglades LTERs), two long-term research sites that are not part of the LTER network (North Inlet and Indian River Lagoon sites), and four experimental sites currently being used by other institutions, including the University of Maine, Fairfield (CT) University, Delaware State University, and East Carolina University. The selected sites represent a gradient of climatic conditions and biogeographic regions (see Table 1) that can be categorized into provinces of similar climate, wave climate, and faunal distribution [30, 31]. The northernmost sites have cold winters (the high temperatures are near or below freezing) and cool summers. The southernmost sites are very warm with average high temperatures near or above 30°C. The latter sites also tend to have the most rainfall, with the bulk of the rain coming in the summer rainy season. Tidal ranges are 1–2 m in most areas except the two Florida sites, which are microtidal. In all cases, flooding of the high marshes is accomplished largely by wind-driven storm tides.

Location of participating sites. Dominant high-marsh plants (standing dead) and sediments were collected from each of the sites in late fall–winter of 2000–2001.
Sampling and Sample Analysis
During the period from November 2000 to January 2001, standing dead plants were cut at each site, placed on ice, and shipped overnight to the University of Virginia where the samples were frozen at −80°C until analysis. Additionally, at the time the plant samples were collected, samples of the sediment were collected by inserting a detipped 10-mL syringe into the sediment to collect a small core that was 1.5 cm × 2 cm. (Sediment samples were not obtained from the Massachusetts site.) The cores were frozen and shipped along with the plant material. Three such cores were collected at each site; two were used for analysis of physical characteristics and for determinations of microbial abundance, and one was used to determine the sediment community structure as a representation of the environment in which the plant materials would eventually decay. At all sites except Virginia, three cores were collected randomly within a 10 m × 3 m plot. Two cores were used to determine sediment moisture content, bulk density, and organic matter concentration. At the Virginia site, 10 cores were collected randomly, but at least 2.0 meters apart, within a 20 × 20 m plot. This spacing exceeds the horizontal distance determined by Franklin et al. [18] with a geostatistical approach to be the spacing necessary to ensure independent samples of the microbial community. Three of the Virginia cores were used to determine sediment moisture content, bulk density, and organic matter content. Seven of the cores were used to determine the variability of the sediment microbial community structure at the site.
Abundance of bacteria and fungi in the plant material and sediments was determined by direct counting methods [5]. Abundance (bacterial and fungal) was estimated for three individual plants and from duplicate sediment cores. Portions (1 g, moist weight) of each plant or sediment sample were placed in a blender and homogenized, and dilutions of each suspension were examined for bacterial abundance by acridine orange direct counts (AODC) using the approach of Rublee and Dornseiff [59]. Fungal abundance was estimated as the hyphal length per gram of dry plant material by the hyphal intersection approach of Jones and Mollison [35]. This method is reported to overestimate the abundance of fungal hyphae because of an accumulation of empty (necrotic) hyphae during decomposition [34]. The magnitude of the bias does not appear to be dependent on the plant identity, and it is relatively small, a factor of about two in decaying Spartina alterniflora [48]. Given the lack of any pattern in the data obtained with this technique (see Fig. 5), conclusions about those results are not likely to be affected by the bias.
To measure the organic content of the plant materials (or sediments), plants were dried at 70°C to a constant mass, weighed, ignited (450°C, overnight), and reweighed. The carbon and nitrogen contents of the plant materials were determined using a PerkinElmer 2400 series II CHNS/O analyzer. C/N was calculated as the ratio of the masses.
Prior to nucleic acid extraction, ~5–10 g of frozen plant tissue was ground to a fine powder in liquid nitrogen using a mortar and pestle. Macerated plant tissue and sediment samples were transferred in triplicate 0.25-g aliquots into 2-mL screw-top tubes for extraction using the UltraClean Soil DNA Isolation Kit (MoBio Laboratories, Solana Beach, CA). DNA was extracted and purified in accordance with the manufacturer’s instructions, as modified by Clement and Kitts [11]. Following extraction, microbial community DNA was amplified by PCR and examined using the terminal restriction fragment length polymorphism (TRFLP) method [42]. As described below, TRFLP of rDNA was used to fingerprint composition within both the bacterial and fungal communities and to cluster each respective community based on shared similarity rather than to assign phylogenetic identities to individual amplicons.
Fungal community PCR was performed in 100-μL reaction volumes using a 6FAM-labeled forward primer (ITS1-F) and a HEX-labeled reverse primer (ITS4) (synthesized by Operon Technologies, Alameda, CA, USA) targeting the intergenic transcribed spacer (ITS) region of the ribosomal DNA operon of the fungi, as described in Klamer et al. [37]. Note that the forward primer is that used by Buchan et al. [7], whereas that group used the ascomycete-targeted reverse primer ITS 4A rather than ITS4 which is reported to target most Ascomycota and Basidiomycota along with some Zygomycota and Chytridiomycota [37]. As shown by Klamer et al. [37], the ITS4 primer amplified Ascomycota with the same efficiency as ITS 4A and also enabled amplification of Basidomycota without nonspecific amplification of plant ITS targets. The primer pair used here has been applied successfully in similar work by Anderson et al. [2].
Bacterial community PCR was performed in 100-μL reaction volumes using a 6FAM-labeled forward primer (Bac-SSU27F) and an unlabeled reverse primer (Bac-SSU1492) targeting the small subunit 16S rDNA region of the ribosomal DNA operon. Primer sequences and optimal annealing temperatures for community PCR in this environment are shown in Table 2. Final PCR reagent concentrations were 2.0 mM MgCl2, 1× Applied Biosystems Buffer II, 200 μM of each dNTP, 1.0 μM of each primer, 0.4 μg/μL BSA (bovine serum albumin), and 1.25 U AmpliTaq DNA polymerase. All amplification reactions were performed in an MJ Research PTC-200 Thermocycler (Waltham, MA, USA) with reagents obtained from Applied Biosystems (Foster City, CA, USA), with the exception of BSA that was obtained from Roche Diagnostics (Indianapolis, IN, USA). Thermocycler reaction conditions for fungal ITS amplification were 5 min initial denaturation at 94°C followed by 35 cycles of 0.5 min at 94°C, 2 min of annealing at 50°C, and 3 min extension at 72°C followed by a final extension step of 5 min at 72°C. Bacterial 16S-rDNA PCR amplifications were identical except that the annealing temperature was 55°C. Reaction yield was determined by 1.5% agarose gel electrophoresis. PCR products were purified and concentrated using a QIAquick PCR Purification Kit (Qiagen, Hilden, Germany), digested with the appropriate restriction endonuclease at 5 U per reaction according to manufacturer’s instructions (New England Biolabs, Beverly, MA, USA). The restriction endonucleases HaeIII and RsaI were used to digest 16S-rDNA targets while the enzyme HinfI was used for digestion of fungal ITS targets. Following restriction, samples were purified and desalted with QIAquick Nucleotide Removal Kit (Qiagen, Hilden, Germany), denatured at 95°C for 10 min, and separated by capillary electrophoresis in an ABI 310 Genetic Analyzer in GeneScan mode (Applied Biosystems, Foster City, CA, USA).
Electropherogram data from the TRFLP analyses were segregated into size classes of two-basepair increments and converted to binary format representing the presence or absence of a terminal restriction fragment (each TRF ± 2 base pairs representing an amplicon). A minimum cutoff of 100 relative fluorescence units was used to discriminate peak signals from background noise. The matrices (amplicon × sample) were examined with principal component analysis (PCA) to visualize differences among the community fingerprints determined for each site and dominant plant type [19, 20, 21, 73]. PCA provides a means of comparing community fingerprints across sites, among plant types, and along environmental gradients. Although PCA ordination is not generally applied to binary data, its use for the visualization of such data where extraction of factors is not attempted is both acceptable and common [16].
Results
Sediment Characteristics
Sediment bulk density, water content, organic matter percentage, and bacterial abundance for the 10 study sites varied widely but were not atypical of marsh sediments (Table 3, Fig. 2). There was no obvious distributional pattern observed in any of the variables measured with respect to latitude, tide range, salinity, or dominant high marsh vegetation; however, organic matter content was positively correlated (r = 0.514) with sediment bacterial abundance (Fig. 2).

Distribution of the abundance of sediment bacteria by latitude (top) and by the percentage of organic matter present in the sediments (bottom).
Sediment bacterial community composition differed from site to site (Fig. 3). At the Virginia site, seven sediment samples were extracted and analyzed to obtain an estimate of the variance associated with the bacterial communities. Error bars representing 1 standard error of the mean (SEM) are included in Fig. 3, indicating that the magnitude of uncertainty associated with these analyses is such that the separation observed among the data points reflects differences among the sites and not with-site variance.

Principal components analysis plot of sediment-bacteria community composition patterns obtained from TRFLP analysis of whole-community DNA extracts amplified with the primer set 27f_6FAM:1492r.
Of the sediment bacterial communities available for analysis, the northernmost sites (Maine and Connecticut) were separated from the rest of the sites on the first PC axis. The second PC axis separates the remainder of the sites along a general latitudinal gradient from Maine to South Florida with samples from the Virginian and Carolinian provinces (Table 1) forming a cluster midway between the Maine and South Florida sites. The exceptions to this pattern are the Virginia and Connecticut sites, which are located outside of that cluster. However, the strength of the relationship between latitude and sediment bacterial community composition is clear when the second PC scores are plotted versus site latitude (Fig. 4, r = 0.583).

Distribution of PC2 values for bacterial sediment community composition (from Fig. 3) with latitude to display latitudinal gradient in community composition of the sediment. The correlation between the variables was r = 0.583.
Microbial Abundance
Plant-associated bacterial abundance roughly corresponded to the latitudinal gradient; there was a general trend of increasing bacterial abundance from the north to south that ranged widely from a high of over 109 to a low of 2 x 106 cells g−1 dry mass of plant material (Fig. 5). If the lack of detectable fungi in the South Florida and Indian River samples is considered, fungal abundance also corresponded to the latitudinal gradient but spanned less than an order of magnitude (Fig. 5). In contrast to bacterial abundance, fungal abundance decreases from north to south. There was no clear effect of plant type on either bacterial or fungal abundance. Although the impact of plant type on microbial abundance may have been confounded by the apparent latitudinal effect, the lack of a detectable plant effect may not be surprising given that similar plant tissues were not necessarily used for microbial enumeration. Instead, the entire plant was ground and used to prepare dilutions so that the proportion of leaves, leaf sheath, and stem varied from sample to sample. As Gessner [23] and Newell [48] note, hyphae are unevenly distributed along standing dead S. alterniflora and Juncus roemerianus leaves. Furthermore, differences in the time between plant senescence and collection of the plant materials used for these analyses may mask the effect of plant type on bacterial and fungal abundance by providing differing lengths of time for standing-dead microbial communities to develop.

Abundance of bacteria (top) and fungi (bottom) in the plant material collected from the 10 sites. There is a data point for bacteria and fungi associated with Spartina alterniflora as samples of this plant from the Virginia site were also included in these analyses. Fungi were below the limit of detection (170 m hyphae g−1 dry wt plant−1) for Spartina bakeri or Rhizophora mangle; data points for the hyphae associate with these two plants represent a point halfway between 0 and the minimum detectable quantity.
Hyphae were not seen in either Rhizophora mangle or Spartina bakeri samples. Given that the minimum detectable quantity of hyphae for the microscopic technique used in this study is about 170 m of hyphae g−1 dry mass of plant material, hyphae could have been present in these samples below this value. For purposes of correlation analysis, a value halfway between 0 and the minimum detectable quantity of hyphae was used for these two sites.
Sediment microbial abundance patterns were different from those for standing dead plants. No fungal hyphae were observed in sediment samples. Bacteria were more abundant in sediments than on standing-dead plant surfaces ranging from a low of 3.9 × 109 cells g−1 dry mass sediment in the North Carolina marsh to over 2 × 1010 cells g−1 dry mass sediment in the Connecticut location (Fig. 2). Sediment bacterial abundance was not correlated with latitude; but it was correlated with the organic matter content of the small cores (1.6 cm diameter × 2 cm long) used for enumeration of the bacteria (Fig. 2).
Plant-Associated Microbial Communities
A distinct difference in plant-associated community composition was observed for both bacteria and fungi (Fig. 6). Bacterial communities associated with Juncus were significantly different on the PC2 axis from the remaining samples which were dominated by three Spartina species (ANOVA, F0.05(8,2) = 7.635, p = 0.014). Phragmites- and Rhizophora-associated bacterial communities clustered along with bacterial communities from the three species of Spartina. The Juncus datum that is distant from the other two Juncus-associated bacterial communities represents vegetation from the Georgia site. This particular microbial community is notable because of the much lower concentration of hyphae on plants from the Georgia site relative to other sites in this study (Fig. 5). The paucity of hyphae on these plant may have influenced the composition of the bacterial community found there or reflect other factors at the Georgia site that affect microbial community composition. The South Carolina S. alterniflora bacterial community was also quite different from the other communities found on Spartina plants. This might be attributable to the noticeable amounts of fine-grained sediments adhering to the plants from the South Carolina site. Substantially less sediment was observed on plants from other sites. Adhering sediments were not removed from plants before extraction of microbial community DNA. It is interesting to note that if the sediment-contaminated South Carolina sample point is excluded from the analysis, the Juncus vs. other plant comparison on the first PC axis is not significant, but is nearly so at p = 0.060 (ANOVA).

Comparison of bacterial and fungal communities from standing dead marsh vegetation representing the six different plants examined. No products were recovered from the amplification of mangrove-associated fungi.
Fungal communities (Fig. 6) from each of the Spartina species alone and in aggregate were more similar to one another than to those from Juncus (which were tightly clustered internally) (ANOVA on PC1, F0.05(5,3) = 7.404, p = 0.027) or Phragmites (a single sample which clustered at a distance from all other plant materials). When the individual Spartina species were entered into the analysis separately (S. bakeri from the Indian River site was deleted as there was only a single representative of that species), the differences were not quite significant for PC1 (p = 0.058), but separation on PC2 was still strongly significant (F0.5(4,3) = 26.36, p = 0.004) suggesting that different fungal communities develop on standing dead leaves of different Spartina species as well as among genera. No amplification products were obtained from Rhizophora communities, consistent with the abundance results. Amplification products were obtained from S. bakeri, although no hyphae were observed in the enumerations. This result might suggest that fungi were present in S. bakeri at a level too low to be seen microscopically. Although the Maine and Connecticut samples did cluster away from the rest of the sites, the lack of overall spatial pattern suggests that plant type affects the structure of the fungal community more than geographic location.
Comparison of Plant and Sediment Bacterial Communities
Sediment bacterial communities were clearly different from plant-associated communities providing evidence that the differences are real community differences among the standing-dead associated microbial communities (Fig. 7). Only one community (South Carolina’s plant-associated community) overlapped with the opposing sediment-associated cluster; however, that sample was very different from its sediment-associated counterpart. This result is consistent with the observation that South Carolina plant materials were coated with sediment particles.

Comparison of plant-associated and sediment-associated bacterial communities showing the distinct differences between the two habitats. Open circles are sediment data; closed circles are plant data.
Discussion
T-RFLP analysis of whole microbial community nucleic acid provides information about the presence of the most abundant microbial amplicons in a community. In our previous experience with the quantification of direct nucleic acid extracts from environmental samples, DNA quantity is not always correlated with amplification efficiency because of the presence of PCR inhibitors coextracted in the purified DNA product. Therefore, nucleic acids were extracted from standardized amounts of plant and sediment materials and then amplified directly rather than adjusting the concentration of DNA used in each T-RFLP analysis. Electropherogram patterns reflect the most abundant amplicons per unit of plant or sediment sample but cannot be used to provide specific information about community richness based on similar numbers of individual organisms (i.e., a standardized amount of DNA template in each reaction tube). Although SSU rDNA-targeted T-RFLP analysis of relatively simple communities can be highly reproducible (e.g., [43]), richness estimates derived from direct extracts and PCR amplification of natural samples are at best qualitative measures because of uncertainties in the biases associated with extraction and amplification efficiency from complex environments [63]. The term community composition is used to refer to community fingerprints throughout the remainder of this article.
Microbial communities from different habitats are indeed different: communities associated with standing-dead plants are distinct from sediment communities (Fig. 7). Although the South Carolina plant-associated microbial community appears to be the single exception to this pattern, the observation that plants from South Carolina were coated with visible amounts of fine sediments while the plants from all other sites did not may explain why this microbial community clusters with sediment communities. The separation of the South Carolina point from the standing-dead communities (Fig. 7) indicates that this community was actually a mixture of bacteria typically associated with standing plants and sediments. It is noteworthy, however, that the South Carolina plant and sediment communities are widely separated along the first PC axis, emphasizing that sediment communities and those on standing-dead plants are different from one another. Like the South Carolina samples, biotic and abiotic conditions at the other nine sites are sufficiently different in these two habitat types that microbial communities on standing-dead marsh plants are discrete from sediment communities even when the habitats compared are in the same physical location in a marsh. The difference between the two habitat types is not surprising given that extraction of community DNA includes anaerobes and chemolithotrophs that might be abundant in the sediment but unlikely to be dominant on standing-dead plant material. It does suggest that when the plant material falls to the sediment surface, the litter will be exposed to an entirely new community of decomposers.
Each habitat type (i.e., plant and sediment) was characterized by distinctly different community patterns along the latitudinal gradient. Within sediments, the bacterial community fingerprints correlate with the latitude differences among the sites (Fig. 3 and Fig. 4), while bacterial and fungal communities associated with standing-dead plants form distinct groups based on plant type (Fig. 6). Sediment community fingerprints were not correlated with sediment bulk density or organic matter content. Plant-associated communities were not correlated with latitude. The community patterns observed in sediments across the wide spatial scales examined here suggest that in some habitats bacterial communities may exhibit biogeographical distribution patterns. What the factor or factors are that covary with latitude and contribute to these biogeographic patterns is not evident from this survey but could include soil type, temperature, precipitation patterns, seasonality, or light intensity.
In the standing-dead plant habitat, latitudinal effects were not detected. Although climatic factors may contribute to the distribution and composition of microbial communities associated with standing-dead marsh plants, plant type is the most likely primary determinant of microbial community composition. Climate effects were of lesser importance than organic substrate impacts on community composition patterns. Organic substrate effects on microbial community processes have been noted previously; salt marsh plants show greater resistance to decay than mangroves [5, 10, 46, 75]. Support for the importance of organic substrate differences affecting community structure of the standing-dead microbial community comes from direct microscopic observation of fungi and the ability to identify the ascomycete fungi from reproductive structures formed in the standing-dead plant tissues. Work with fungal communities on Phragmites australis (K.D. Hyde, personal communication) and the salt marsh plants S. alterniflora and J. roemerianus [38, 39] clearly shows that there is very little or no overlap among the decomposition communities of ascomycete fungi on different species of marsh grass. Newell’s microscopic work is consistent with the rDNA ITS results of Buchan et al. [7] in that all but a couple of minor T-RFLP peaks from naturally decaying smooth-cordgrass blades match to their set of directly observed ascomycetes. Several of the species (especially rarer ones) observed directly have not shown up in the T-RFLP scans, including the ascomycete yeasts that have been reported from smooth cordgrass. Also, the lack of amplification with fungal primers in R. mangle leaves is consistent with Newell’s findings, at least for leaves decaying in persistently submerged situations. The primary eukaryotic decomposers of mangrove leaves are not true fungi but mycelial straminipiles (oomycotes, species of Halophytophthora) that may not be detected with the primers used here [6, 7].
If organic substrates, determined by plant type, have such a strong influence on standing dead microbial communities, why then don’t sediment communities segregate by the type of marsh plant contributing the greatest proportion of the sediment carbon or by sediment organic matter content? Perhaps the answer is that salt marsh sediments along the East Coast are more chemically homogenous environments than standing-dead plants so that climate factors such as temperature, or sediment edaphic factors, are the variables that differ the most among the locations we examined. By the time standing-dead plants are deposited on the sediment surface as litter, the litter has been decayed to such an extent that any differences in the living (or recently dead) plant materials have been eliminated. The lack of community composition differences and the positive relationship between sediment organic matter content and bacterial abundance support this hypothesis.
The positive correlation (Fig. 2, r = 0.514) between sediment organic matter and bacterial abundance suggests that sediments rich in organic matter have a higher microbial biomass carrying capacity than low organic matter sediments. Fungi, on the other hand were generally rare in the sediments sampled. The link between organic carbon and bacterial abundance is well established in soils, sediments, and water [1] and has been attributed to a general carbon limitation of bacteria in environmental samples [1]. The absence of fungal hyphae in the marsh sediments examined in this study is not remarkable, as fungi are generally not found in wetland soils [27], although live, active fungi have been observed in surface peat of fresh [27] and salt water marshes [44] or associated with live plant roots as mycorrhizae [13, 14, 32, 68, 72]. Several reports [26, 27, 51] suggest that fungi have the potential to play a significant role in sediment organic matter decomposition, but reports such as these are limited; the distribution and role of fungi in a wider range of (frequently anoxic) sediments remain unclear.
Bacterial and fungal abundance were correlated with latitude (Fig. 5). The effect is more pronounced for bacteria than for fungi and the correlations are opposite to one another: bacterial abundance decreased with increasing latitude while fungal hyphae became more abundant as latitude increased. Gradients in microbial abundance have been noted previously for bacteria and have been attributed to temperature effects (e.g., [36]). Newell [47] and Samiaji and Bärlocker [60] hypothesized that fungal biomass associated with standing-dead marsh grasses should increase with decreasing latitude, but the present results displayed the opposite effect. Furthermore, these results differ from other literature reports. In a survey of standing-dead S. alterniflora collected at a similar time of year from sites ranging from northern Florida to Maine, Newell et al. [49] did not detect a gradient in fungal biomass or activity in the 3 years samples were collected. In that report, 21% of the variation in fungal biomass could be attributed to variation in the C/N ratio of the standing-dead material (the higher the C/N ratio, the higher the fungal biomass). Similar results were not observed for the plant materials used in our study: the relationship between C/N ratio and microbial abundance (hyphal or bacterial) was opposite to that observed by Newell et al. [49], but the correlation was not significant and could explain only 11% of the variation in abundance. The abundance patterns we report are confounded by differences in the plant species collected; however, even when fungi associated with Spartina alterniflora or all examined Spartina species are considered (Fig. 5), the patterns are still in opposition to that of Newell et al. [49]. The difference in the patterns between these two studies may have resulted from the difference in the techniques used to measure fungal abundance. Newell et al. [49] based their measurements on ergosterol, which is most likely to be associated with living fungi [69]. Ergosterol content may, however, more accurately reflect changes in species composition rather than changes in fungal abundance in some environments [4, 52]. Fungal abundance estimates reported here are based on microscopic examination of fungal hyphae, a method that can overestimate the abundance of living fungi because empty hyphae cannot be distinguished from living hyphae [34, 48].
The inverse relationship between bacteria and fungal abundance (Fig. 5) suggests the potential for a competitive or antagonistic relationship between bacteria and fungi: as the abundance of one of these types of microbes increases, the other decreases. However, limitations of the microscopic techniques used, as noted above for the fungi, make comparisons of bacterial to fungal biomass unreliable. Newell [47] hypothesized that a mutualistic relationship exists between bacteria and fungi on standing dead plants in which fungi create a more hospitable environment for the growth of bacteria citing, for example, Wilson et al. [74], who show that fungi metabolize S. alterniflora cinnamic acid, an antiherbivory compound that also exhibits antimicrobial activity. To test Newell’s hypothesis about the relationship between bacteria and fungi, measurement of living biomass and metabolic activity would provide greater insight into the nature of the microbial interactions on standing-dead plants than the methods used in our study.
Two of the plant types examined supported very low or no fungal populations (Fig. 5, bottom). The lack of detectable fungi on yellow-but-attached red mangrove leaves by microscopic methods suggests that hyphal fungi were not abundant or were not present on the mangrove leaves. Our inability to amplify fungal DNA from these samples, microscopic examination by other investigators [5], and quantification of ergosterol [5, 50, 55, 56] in red mangrove leaves support the observation that hyphal fungi are not found in association with R. mangle leaves. It is important to note that polysaccharides, polyphenolics, which are relatively abundant in sediments, and many leaf tissues including red mangrove, can interfere with PCR amplification [61, 66, 70] and that other investigators have attributed an inability to amplify DNA from similar plant samples to this technical problem [53]. However, our ability to amplify bacterial DNA from these same red mangrove extracts suggests that the DNA used in the PCR reactions was sufficiently purified to avoid polyphenolic inhibition of DNA amplification during PCR. It is possible that the amount of fungal DNA in the extracts was simply below the level of amplification by PCR. Unlike the red-mangrove case, T-RFLP analysis of S. bakerii plants revealed that fungi were present in the plant materials below the limit of microscopic detection even though no fungal hyphae were found during microscopic examination. This leads to the conclusion that fungi may be relatively less important in decay of standing dead S. bakerii than the abundant bacteria associated with this plant—a conclusion that is counter to extensive evidence that fungi are critical to decomposition of a wide variety of standing-dead marsh plants. Confirmation of this conclusion would require more extensive study of S. bakerii than examination of the three plant leaves and stems included in this study.
There is a strong biogeographical component to the relative abundance of bacterial and fungal communities associated with standing-dead plant biomass, but microbial community composition on standing-dead plants is not strongly influenced by the latitudinal gradient. Conversely, bacterial abundance in the sediments was not associated with latitude but was clearly correlated with sediment organic matter content. Within the sediment, the composition of both bacterial and fungal communities was strongly correlated with latitude, indicating that biogeographical factors are important determinants of sediment community composition. The composition of both the fungal and the bacterial communities on standing-dead plants was more similar among plants of the same type (i.e., related plant species) indicating that plant type (and hence substrate quality) are primarily responsible for the microbial community composition there. The colonization of standing-dead plants is similar in this respect to the colonization of the rhizosphere where there appears to be strong selection for particular microbial types [40]. Although no correlation was evident between latitude and community composition for standing-dead microbial communities, both bacterial and fungal abundance on standing-dead plants varied with latitude.
Whereas bacterial abundance on standing-dead plants and latitude were positively correlated, fungal abundance on standing-dead plants increased with decreasing latitude. Relative microbial abundance on standing-dead plants in the salt marsh is therefore clearly determined by factors relating to the biogeography of the environment. If the bacterial and fungal community composition were considered together, the inverse relationship in abundance might show a latitudinal effect on overall microbial community composition. We have begun to address this question as part of the evaluation of the community succession during decay.
The observed differences in sediment community composition across sites means that different microbial inocula may be available to colonize standing-dead plant material after it falls to the sediment surface and continues the decay process there. If the developing microbial decomposition community is a combination of the resident standing-dead and sediment communities, the mature decay communities may be substantially different among sites even though the fundamental decay process is similar among sites. The balance of natural selection and recruitment in these mature decomposition communities may be strongly influenced by the biogeography of the environment. It will be interesting to observe if the biogeographical component of sediment microbial composition is maintained within the decomposition communities or if its signature is lost through time. Decomposition experiments currently in progress at the 10 sites will enable resolution between these potential ecological outcomes.
References
M Alexander (1977) Introduction to Soil Microbiology, 2nd ed. Wiley New York
IC Anderson CD Campbell JI Prosser (2003) ArticleTitlePotential bias of fungal 18S rDNA and internal transcribed spacer polymerase chain reaction primers for estimating fungal biodiversity in soil. Environ Microbiol 5 36–47 Occurrence Handle10.1046/j.1462-2920.2003.00383.x Occurrence Handle1:CAS:528:DC%2BD3sXhs1WntbY%3D Occurrence Handle12542711
Baas Becking, LGM (1934) Geobiologie of inleiding tot de milieukunde Diligentia Wetensch. van Stockum’s, Gravenhage
S Bermingham L Maltby RC Cooke (1995) ArticleTitleA critical assessment of the validity of ergosterol as an indicator of fungal biomass. Mycol Res 99 479–484 Occurrence Handle1:CAS:528:DyaK2MXlvFCksr4%3D
LK Blum AL Mills JC Zieman RT Zieman (1988) ArticleTitleAbundance of bacteria and fungi in seagrass and mangrove detritus. Mar Ecol Prog Ser 42 73–78
P Bridge B Spooner (2001) ArticleTitleSoil fungi: diversity and detection. Plant Soil 232 147–154 Occurrence Handle10.1023/A:1010346305799 Occurrence Handle1:CAS:528:DC%2BD3MXlsVCksL0%3D
A Buchan SY Newell JIL Moreta MA Moran (2002) ArticleTitleAnalysis of internal transcribed spacer (ITS) regions of rRNA genes in fungal communities in a southeastern US salt marsh. Microb Ecol 43 329–340 Occurrence Handle10.1007/s00248-001-1062-0 Occurrence Handle1:CAS:528:DC%2BD38XkvFSnsro%3D Occurrence Handle12037611
FL Bunnell DEN Tait PW Flanagan K van Cleave (1977) ArticleTitleMicrobial respiration and substrate loss. I. A general model of the influences of abiotic factors. Soil Biol Biochem 9 33–40 Occurrence Handle10.1016/0038-0717(77)90058-X Occurrence Handle1:CAS:528:DyaE2sXlt1Gltg%3D%3D
FL Bunnell EEN Tait (1977) ArticleTitleMicrobial respiration and substrate loss. II. A model of the influences of chemical composition. Soil Biol Biochem 9 41–47 Occurrence Handle10.1016/0038-0717(77)90059-1 Occurrence Handle1:CAS:528:DyaE2sXlsVakug%3D%3D
RR Christian (1989) Microbial ecology and organic detritus in estuaries. A Yáñez-Arancibia (Eds) Estuarine Ecology WiIey New York
BG Clement CL Kitts (2000) ArticleTitleIsolating PCR-quality DNA from human feces with a soil DNA kit. Biotechniques 28 640 Occurrence Handle1:CAS:528:DC%2BD3cXis1Wmurw%3D Occurrence Handle10769741
SA Connon SJ Giovannoni (2002) ArticleTitleHigh-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl Environ Microbiol 68 3878–3885 Occurrence Handle10.1128/AEM.68.8.3878-3885.2002 Occurrence Handle1:CAS:528:DC%2BD38XmtVaqu7o%3D Occurrence Handle12147485
JC Cooke RH Butler G Madole (1993) ArticleTitleSome observations on the vertical-distribution of vesicular-arbuscular mycorrhizae in roots of salt-marsh grasses growing in saturated soils. Mycologia 85 547–550
JC Cooke MW Lefor (1990) ArticleTitleComparison of vesicular arbuscular mycorrhizae in plants from disturbed and adjacent undisturbed regions of a coastal salt-marsh in Clinton, Connecticut, USA. Environmental Management 14 131–137
TP Curtis WT Sloan JW Scannel (2002) ArticleTitleEstimating prokaryotic diversity and its limits. Proc Nat Acad Sci USA 99 10491–10499 Occurrence Handle10.1073/pnas.142680199
SL Dollhopf SA Hashsham JM Tiedje (2001) ArticleTitleInterpreting 16S rDNA T-RFLP data: application of self-organizing maps and principal component analysis to describe community dynamics and convergence. Microb Ecol 42 495–505 Occurrence Handle10.1007/s00248-001-0027-7 Occurrence Handle1:CAS:528:DC%2BD38XntFWrtg%3D%3D Occurrence Handle12024232
GM Edelman JA Gally (2001) ArticleTitleDegeneracy and complexity in biological systems. Proc Nat Acad Sci USA 98 13763–13768 Occurrence Handle10.1073/pnas.231499798 Occurrence Handle1:CAS:528:DC%2BD3MXovVynt74%3D Occurrence Handle11698650
RB Franklin LK Blum A McComb AL Mills (2002) ArticleTitleA geostatistical analysis of small-scale spatial variability in bacterial abundance and community structure in salt-marsh creek bank sediments. FEMS Microbiol Ecol 42 71–80 Occurrence Handle10.1016/S0168-6496(02)00320-3 Occurrence Handle1:CAS:528:DC%2BD38Xnt1KmtL4%3D Occurrence Handle12542032
RB Franklin JL Garland CH Bolster AL Mills (2001) ArticleTitleThe impact of dilution on microbial community structure and functional potential: a comparison of numerical simulations and batch culture experiments. Appl Environ Microbiol 67 702–712 Occurrence Handle10.1128/AEM.67.2.702-712.2001 Occurrence Handle1:CAS:528:DC%2BD3MXhtFSlsrw%3D Occurrence Handle11157234
RB Franklin AL Mills (2003) ArticleTitleMulti-scale variation in spatial heterogeneity of microbial community structure in an eastern Virginia agricultural field. FEMS Microbiol Ecol 44 IssueID3 335–46 Occurrence Handle10.1016/S0168-6496(03)00074-6 Occurrence Handle1:CAS:528:DC%2BD3sXjs1Ckurk%3D Occurrence Handle12830827
RB Franklin DR Taylor AL Mills (1999) ArticleTitleCharacterization of microbial communities using randomly amplified polymorphic DNA (RAPD). J Microbiol Methods 35 225–235 Occurrence Handle10.1016/S0167-7012(99)00003-2 Occurrence Handle1:CAS:528:DyaK1MXitFant7k%3D Occurrence Handle10333074
M Gardes TD Bruns (1993) ArticleTitleITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol Ecol 2 113–118 Occurrence Handle1:CAS:528:DyaK3sXlslOmsro%3D Occurrence Handle8180733
RV Gessner (1977) ArticleTitleSeasonal occurrence and distribution of fungi associated with Spartina alterniflora from a Rhode Island estuary. Mycologia 69 477–491
MS Girvan J Bullimore JN Pretty AM Osborn AS Ball (2003) ArticleTitleSoil type is the primary determinant of the composition of the total and active bacterial communities in arable soils. Appl Environ Microbiol 69 1800–1809 Occurrence Handle10.1128/AEM.69.3.1800-1809.2003 Occurrence Handle1:CAS:528:DC%2BD3sXitlCltrY%3D Occurrence Handle12620873
GL Godshalk RG Wetzel (1978) ArticleTitleDecomposition of aquatic angiosperms. II. Particulate components. Aquat Bot 5 301–327 Occurrence Handle10.1016/0304-3770(78)90074-8 Occurrence Handle1:CAS:528:DyaE1MXhslCgurk%3D
CT Hackney (1987) ArticleTitleFactors affecting accumulation or loss of macroorganic matter in salt marsh sediments. Ecology 68 1109–1113
CT Hackney DE Padgett MH Posey (2000) ArticleTitleFungal and bacterial contributions to the decomposition of Cladium and Typha leaves in nutrient enriched and nutrient poor areas of the Everglades, with a note on ergosterol concentrations in Everglades soils. Mycol Res 104 666–670 Occurrence Handle10.1017/S0953756299002221
Å Hagström T Pommier F Rohwer K Simu W Stolte D Svensson UL Zweifel (2002) ArticleTitleUse of 16S ribosomal DNA for delineation of marine bacterioplankton species. Appl Environ Microbiol 68 3628–3633 Occurrence Handle10.1128/AEM.68.7.3628-3633.2002 Occurrence Handle12089052
EB Haines RB Hanson (1979) ArticleTitleExperimental degradation of detritus made from the salt marsh plants Spartina alterniflora Loisel, Salicornia virginica L., and Juncus roemerianus Scheele. J Exp Mar Biol Ecol 40 27–40 Occurrence Handle10.1016/0022-0981(79)90031-5 Occurrence Handle1:CAS:528:DyaE1MXlsFKltbg%3D
BP Hayden R Dolan (1973) Classification of the coastal environments of the world Rep. No. AD/A-008-578-FWN, University of Virginia Charlottesville, Virginia
BP Hayden R Dolan (1976) ArticleTitleCoastal marine fauna and marine climates of the Americas. J Biogeog 3 71–81
MH Hoefnagels SW Broome SR Shafer (1993) ArticleTitleVesicular–arbuscular mycorrhizae in salt marshes in North Carolina. Estuaries 16 851–858
PE Jansson B Berg (1985) ArticleTitleTemporal variation of litter decomposition in relation to simulated soil climate: long-term decopositon in a Scots pine forest. Can J Bot 63 1008–1016
RG Joergensen B Meyer A Roden B Wittke (1996) ArticleTitleMicrobial activity and biomass in mixture treatments of soil and biogenic municipal refuse compost. Biol Fertil Soils 23 43–49 Occurrence Handle10.1007/s003740050136
JG Jones JE Mollison (1948) ArticleTitleA technique for the quantitative estimation of soil microorganisms. J Gen Microbiol 2 54–69 Occurrence Handle1:CAS:528:DyaH1cXjtVCntA%3D%3D
J Karlsson A Jonsson M Jansson (2001) ArticleTitleBacterioplankton production in lakes along an altitude gradient in the subarctic north of Sweden. Microb Ecol 42 372–382 Occurrence Handle10.1007/s00248-001-0009-9 Occurrence Handle1:CAS:528:DC%2BD38Xks1I%3D Occurrence Handle12024262
M Klamer MS Roberts LH Levine BG Drake JL Garland (2002) ArticleTitleInfluence of elevated carbon dioxide on the fungal community in a coastal scrub oak forest soil investigated using terminal restriction length polymorphism analysis. Appl Environ Microbiol 68 IssueID9 4370–6 Occurrence Handle10.1128/AEM.68.9.4370-4376.2002 Occurrence Handle1:CAS:528:DC%2BD38XmvVequ7k%3D Occurrence Handle12200289
J Kohlmeyer B Volkmann-Kohlmeyer (2001) ArticleTitleThe biodiversity of fungi on Juncus roemerianus. Mycol Res 105 1411–1412
J Kohlmeyer B Volkmann-Kohlmeyer (2002) ArticleTitleFungi on Juncus and Spartina New marine species of Anthostomella: with a list of marine fungi known on Spartina. Mycol Res 106 365–374 Occurrence Handle10.1017/S0953756201005469
GA Kowalchuk DS Buma W de Boer PGL Klinkhamer JA van Veen (2002) ArticleTitleEffects of above-ground plant species composition and diversity on the diversity of soil-borne microorganisms. Antonie van Leeuwenhoek 81 509–520 Occurrence Handle10.1023/A:1020565523615 Occurrence Handle12448746
DJ Lane (1991) 16S/23S Sequencing. E Stackebrandt M Goodfellow (Eds) Nucleic Acid Techniques in Bacterial Systematics Wiley New York 115–176
W-T Liu TL Marsh H Cheng LJ Forney (1997) ArticleTitleCharacterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl Environ Microbiol 63 4516–4522 Occurrence Handle1:CAS:528:DyaK2sXnt12ntbs%3D Occurrence Handle9361437
T Lueders MW Friedrich (2003) ArticleTitleEvaluation of PCR amplification bias by terminal restriction fragment length polymorphism analysis of small-subunit rRNA and mcrA genes by using defined template mixtures of methanogenic pure cultures and soil DNA extracts. Appl Environ Microbiol 69 320–326 Occurrence Handle10.1128/AEM.69.1.320-326.2003 Occurrence Handle1:CAS:528:DC%2BD3sXkvValsw%3D%3D Occurrence Handle12514011
SD Mansfield F Barlocher (1993) ArticleTitleSeasonal variation of fungal biomass in the sediment of a salt marsh in New Brunswick. Microb Ecol 26 37–45
V Meentenmeyer (1978) ArticleTitleMacroclimate and lignin control of litter decomposition rates. Ecology 59 465–472 Occurrence Handle1:CAS:528:DyaE1cXmtFCht7c%3D
SY Newell (1984) ArticleTitleBacterial and fungal productivity in the marine environment: A contrastive overview. Colloq Int Cent Natl Rech Sci 331 133–139
SY Newell (1993) ArticleTitleDecomposition of shoots of a salt-marsh grass; methodology and dynamics of microbial assemblages. Adv Microb Ecol 13 301–326
SY Newell (1996) ArticleTitleEstablished and potential impacts of eukaryotic mycelial decomposers in marine/terrestrial ecotones. J Exp Mar Biol Ecol 200 187–206 Occurrence Handle10.1016/S0022-0981(96)02643-3
SY Newell LK Blum RE Crawford T Dai M Dionne (2000) ArticleTitleAutumnal biomass and potential productivity of salt marsh fungi from 29° to 43° north latitude along the United States Atlantic coast. Appl Environ Microbiol 66 180–185 Occurrence Handle1:CAS:528:DC%2BD3cXktlSmtw%3D%3D Occurrence Handle10618221
SY Newell JW Fell (1992) ArticleTitleErgosterol content of living and submerged, decaying leaves and twigs of red mangrove. Can J Microbiol 38 979–982 Occurrence Handle1:CAS:528:DyaK3sXhvFantg%3D%3D
DE Padgett DA Celio JH Hearth CT Hackney (1989) ArticleTitleGrowth of filamentous fungi in a surface-sealed woody substratum buried in salt-marsh sediments. Estuaries 12 142–144
DE Padgett MH Posey (1993) ArticleTitleAn evaluation of the efficiencies of several ergosterol extraction techniques. Mycol Res 97 1476–1480 Occurrence Handle1:CAS:528:DyaK2cXjtFyltbY%3D
RM Pandey RP Adams LE Flournoy (1996) ArticleTitleInhibition of random amplified polymorphic DNAs (RAPDS) by plant polysaccharides. Plant Mol Biol Reporter 14 17 Occurrence Handle1:CAS:528:DyaK28XislKns7s%3D
A Quispel (1998) ArticleTitleLourens G.M. Baas Becking (1895–1963), inspirator for many (micro)biologists. Int Microbiol 1 69–72 Occurrence Handle1:STN:280:DC%2BD3cvhtFOruw%3D%3D Occurrence Handle10943344
S Raghukumar V Sathe-Pathak S Sharma C Raghukumar (1995) ArticleTitleThraustochytrid and fungal component of marine detritus. III. Field studies on decomposition of leaves of the mangrove Rhizophora apiculata. Aquat Microb Ecol 9 117–125
S Raghukumar S Sharma C Raghukumar V Sathe-Pathak D Chandramohan (1994) ArticleTitleThraustochytrid and fungal component of marine detritus. IV. Laboratory studies on decomposition of leaves of the mangrove Rhizophora apiculata Blume. J Exp Mar Biol Ecol 183 113–131 Occurrence Handle10.1016/0022-0981(94)90160-0
DL Rice (1982) ArticleTitleThe detritus nitrogen problem: new observations and perspectives from organic geochemistry. Mar Ecol Prog Ser 9 153–162 Occurrence Handle1:CAS:528:DyaL38XlsVers7c%3D
MS Roberts JL Garland AL Mills (2004) ArticleTitle Microbial astronauts: assembling microbial communities for advanced life support systems. Microb Ecol 47 IssueID2 137–49 Occurrence Handle10.1007/s00248-003-1060-5 Occurrence Handle1:STN:280:DC%2BD2c3lslGmug%3D%3D Occurrence Handle14994179
P Rublee BE Dornseif (1978) ArticleTitleDirect counts of bacteria in the sediments of a North Carolina salt marsh. Estuaries 1 188–191
J Samiaji F Bärlocher (1996) ArticleTitleGeratology and decomposition of spartina alterniflora Loisel in a New Brunswick saltmarsh. J Exp Mar Biol Ecology 201 233–252 Occurrence Handle10.1016/0022-0981(95)00211-1
RJ Steffan J Goksoyr AK Bej RM Atlas (1988) ArticleTitleRecovery of DNA from soils and sediments. Appl Environ Microbiol 54 2908–2915 Occurrence Handle1:CAS:528:DyaL1MXlsVSrtA%3D%3D Occurrence Handle2851961
V Stuart MI Lucas RC Newell (1981) ArticleTitleHeterotrophic utilization of particulate matter from the kelp Laminaria pallida. Mar Ecol Prog Ser 4 337–348
MT Suzuki SJ Giovannoni (1997) ArticleTitleBias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62 625–630
MJ Swift OW Heal JM Anderson (1979) Decomposition in Terrestrial Ecosystems University of California Press Los Angeles
KR Tenore (1983) ArticleTitleWhat controls the availability to animals of detritus derived from vascular plants: organic nitrogen enrichment or caloric availability? Mar Ecol Prog Ser 10 307–309
YL Tsai BH Olson (1992) ArticleTitleRapid method for separation of bacterial DNA from humic substances in sediments for polymerase chain reaction. Appl Environ Microbiol 58 2292–2295 Occurrence Handle1:CAS:528:DyaK38XltVGhtbg%3D Occurrence Handle1386212
I Valiela J Wilson R Buschbaum C Rietsma D Bryant K Foreman J Teal (1984) ArticleTitleImportance of chemical composition of salt marsh litter on decay rates and feeding by detritivores. Bull Mar Sci 35 261–269
WE Vanduin J Rozema WHO Ernst (1990) ArticleTitleSeasonal and spatial variation in the occurrence of vesicular–arbuscular (VA) mycorrhiza in salt-marsh plants. Agric Ecosys Environ 29 107–110 Occurrence Handle10.1016/0167-8809(90)90263-D
AW West WD Grant GP Spalding (1987) ArticleTitleUse of ergosterol, diminopimelic acid and glucosamine content of soils to monitor changes in microbial populations. Soil Biol Biochem 19 607–612 Occurrence Handle10.1016/0038-0717(87)90106-4 Occurrence Handle1:CAS:528:DyaL2sXlsFegur0%3D
TJ White T Bruns S Lee JW Taylor (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. MA Innis DH Gelfand JJ Sninsky TJ White (Eds) PCR Protocols: A Guide to Methods and Applications Academic Press San Diego 315–322
WB Whitman DC Coleman WJ Wiebe (1998) ArticleTitleProkaryotes: the unseen majority. Proc Nat Acad Sci USA 95 6578–6583 Occurrence Handle10.1073/pnas.95.12.6578 Occurrence Handle1:CAS:528:DyaK1cXjslyntro%3D Occurrence Handle9618454
C Wigand JC Stevenson (1994) ArticleTitleThe presence and possible ecological significance of mycorrhizae of the submersed macrophyte, Vallisneria americana. Estuaries 17 206–215
P Wikstrom A-C Andersson M Forsman (1999) ArticleTitleBiomonitoring complex microbial communities using random amplified polymorphic DNA and principal component analysis. FEMS Microbiol Ecol 28 131 Occurrence Handle10.1016/S0168-6496(98)00098-1 Occurrence Handle1:CAS:528:DyaK1MXhvFCrsbc%3D
JO Wilson R Buschsbaum I Valiela T Swain (1986) ArticleTitleDecomposition in salt marsh ecosystems: phenolic dynamics during decay of litter of Spartina alterniflora. Mar Ecol Prog Ser 29 177–187
CJ Zieman (1975) Quantitative and dynamic aspects of the ecology of turtle grass, Thalassia testudinum. LE Cronin (Eds) Estuarine Research, vol. 1 Academic Press New York 541–562
Acknowledgments
Gary King, University of Maine; Chuck Hopkinson, Marine Biological Laboratory, PIE-LTER; Randy Chambers, Fairfield University; Michael Reiter, Delaware State University; Robert Christian, East Carolina University; James Morris, University of South Carolina; Steven Newell, University of Georgia, GCE-LTER; and Joseph Boyer, Florida International University, ECE-LTER. Technical assistance was provided by Jennifer Adams and Greg Harp. Support for the project was provided under an LTER Intersite Comparison Grant (DEB-0087256) from the NSF.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Blum, L.K., Roberts, M., Garland, J. et al. Distribution of Microbial Communities Associated with the Dominant High Marsh Plants and Sediments of the United States East Coast. Microb Ecol 48, 375–388 (2004). https://doi.org/10.1007/s00248-003-1051-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-003-1051-6