
Abstract
Autoinflammatory diseases constitute a family of disorders defined by aberrant stimulation of inflammatory pathways without involving antigen-directed autoimmunity. They may be divided into monogenic and polygenic types. Monogenic autoinflammatory syndromes are those with identified genetic mutations, such as familial Mediterranean fever, tumor necrosis factor receptor-associated periodic fever syndrome (TRAPS), mevalonate kinase deficiency or hyperimmunoglobulin D syndrome, cryopyrin-associated periodic fever syndromes (CAPS), pyogenic arthritis pyoderma gangrenosum and acne (PAPA) syndrome, interleukin-10 and interleukin-10 receptor deficiencies, adenosine deaminase 2 deficiency and pediatric sarcoidosis. Those without an identified genetic mutation are known as polygenic and include systemic-onset juvenile idiopathic arthritis, idiopathic recurrent acute pericarditis, Behçet syndrome, chronic recurrent multifocal osteomyelitis and inflammatory bowel disease among others. Autoinflammatory disorders are defined by repeating episodes or persistent fever, rash, serositis, lymphadenopathy, arthritis and increased acute phase reactants, and thus may mimic infections clinically. Most monogenic autoinflammatory syndromes present in childhood. However, because of their infrequency, diverse and nonspecific presentation, and the relatively new genetic recognition, diagnosis is usually delayed. In this article, which is Part 1 of a two-part series, the authors update monogenic autoinflammatory diseases in children with special emphasis on imaging features that may help establish the correct diagnosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The term autoinflammation was originally used to differentiate autoimmune disorders from diseases with no specific autoantibodies or autoreactive lymphocytes that were thought to be induced by innate rather than adaptive immune dysregulation [1, 2]. However, we now know that there is overlap in the immunological characteristics of some of the autoinflammatory diseases with the classical autoimmune disorders [2].
Autoinflammatory diseases are further divided into monogenic and polygenic autoinflammatory syndromes, depending on whether there is an identified genetic mutation. Monogenic autoinflammatory syndromes are those with identified genetic mutations and represent, for the most part, abnormal responses of innate immunity. This group includes disorders such as familial Mediterranean fever, tumor necrosis factor receptor-associated periodic syndrome (TRAPS), hyperimmunoglobulin D with periodic fever syndrome or mevalonate kinase deficiency, cryopyrin-associated periodic fever syndromes (CAPS), pyogenic arthritis pyoderma gangrenosum and acne (PAPA) syndrome, interleukin-10 and interleukin-10 receptor deficiencies, adenosine deaminase 2 deficiency and pediatric sarcoidosis. Those without an identified genetic mutation have a polygenic origin and include systemic-onset juvenile idiopathic arthritis, idiopathic recurrent acute pericarditis, Behçet syndrome, chronic recurrent multifocal osteomyelitis and inflammatory bowel disease, among others [3].
An autoinflammatory disorder should be considered in children with recurrent or persistent systemic inflammation that is not explained by other causes, such as infection or malignancy. In most cases, clinical manifestations first appear in childhood. These may include fever, rash, serositis, arthritis, meningitis, uveitis, lymphadenopathy and splenomegaly. Secondary amyloidosis can complicate long-standing disease but is exceptionally rare in childhood. Acute phase reactants are increased during disease flares and may also be abnormal between episodes. Because of the infrequency of autoinflammatory diseases, their diverse and nonspecific presentation, and the relatively new genetic recognition, diagnosis is usually delayed.
In Part 1 of this series, we review monogenic autoinflammatory diseases, focusing on entities that are more pertinent to pediatric radiologists. Although we highlight the most relevant imaging features that may help establish the correct diagnosis, most of the imaging findings are nonspecific and categorization into a definite entity is not always possible. However, delineating organ involvement is more important than diagnostic labels and imaging examinations are particularly useful in this regard.
Periodic fever syndromes
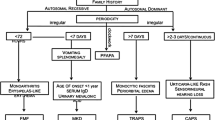
Fever is a frequent flag for sickness in children and is usually caused by infection. However, once infection has been excluded, rheumatological diseases and malignancy should be considered. Relatively frequent peaks of fever of unknown origin combined with specific symptoms are classified as recurrent or periodic fever syndromes, which are listed in Table 1.
Familial Mediterranean fever
Familial Mediterranean fever is the most common monogenic autoinflammatory disorder [4]. It has an autosomal recessive pattern of inheritance, due to genetic alterations in the MEFV gene located on chromosome 16 [4, 5]. MEFV generates the protein pyrin, which is a mediator implicated in several autoinflammatory disorders that participates in the innate immune system and regulates the production of interleukin-1β [6].
Familial Mediterranean fever is more common among non-Ashkenazi Jews, Arabs, Armenians and Turks [6]. The diagnosis is made on the basis of recurrent brief episodes of fever and abdominal, thoracic, articular or cutaneous manifestations with no recognizable infection. Mediterranean origin, positive family history, beginning of the symptoms in childhood or adolescence and good response to colchicine favor the diagnosis. Furthermore, genetic analysis is an additional useful diagnostic tool.
The main cause of painful recurrent abdominal attacks in familial Mediterranean fever is peritonitis, which is a nonspecific presentation and more frequently secondary to other causes of acute abdomen that need to be excluded before familial Mediterranean fever is considered in the differential diagnosis (Table 2). Abdominal imaging findings include signs of paralytic ileus, hepatosplenomegaly, ascites, mesenteric/peritoneal thickening, retroperitoneal inflammation and fibrosis, and lymphadenopathy, including retroperitoneal lymphadenopathy (Figs. 1, 2, 3) [7,8,9]. Bowel wall thickening is also seen occasionally and is believed to be secondary to focal peritonitis. It may also be due to familial Mediterranean fever-associated diseases such as inflammatory bowel disease and Henoch-Schönlein purpura, as well as to secondary amyloidosis, which is extremely rare in children because it only occurs after long-standing inflammation [10].

Familial Mediterranean fever in a 2-year-old girl with abdominal pain for 2 months. Axial contrast-enhanced CT image of the abdomen shows diffuse dilatation of small and large bowel loops with air-fluid levels in keeping with paralytic ileus (arrows). Lack of a transition point helped in the differentiation from small bowel obstruction. There were no inflammatory changes in the remainder of the abdomen

Familial Mediterranean fever in a 6-year-old girl with fever and abdominal pain. Transverse abdominal sonogram of the abdomen shows lymphadenopathy (asterisk), as well as thickening, hyperechogenicity and hyperemia of the mesentery (arrow), associated with a small amount of free fluid (arrowhead). In this setting, differential diagnosis includes other causes of acute abdomen, such as acute appendicitis

Familial Mediterranean fever in a 16-year-old girl with abdominal pain and vomiting. Axial (a) and coronal (b) contrast-enhanced CT images of the abdomen show a small amount of fluid and stranding of the fat in the retroperitoneum extending into the perinephric fat and into the paracolic gutters bilaterally in keeping with inflammatory changes in the retroperitoneum (arrows). Note also splenomegaly (arrowhead in b). Although rare in children, the differential diagnosis includes retroperitoneal fibrosis, which presents with fibrous tissue that envelops the great vessels and ureters with associated hydronephrosis, which can be idiopathic or secondary to prior surgery, malignancy, infection, autoimmune diseases (such as systemic lupus erythematosus or juvenile idiopathic arthritis) and drugs
Chest pain may be due to inflammation of the pleura or referred pain from subdiaphragmatic inflammation. Chest radiographs usually show unilateral pleural effusion with or without adjacent air space disease [11]. Concomitant pericarditis with pericardial thickening and effusion can also be found [12].
Articular symptoms are frequent and may be the initial evidence of disease in pediatric patients [13]. Arthritis is commonly nonerosive, mono- or oligoarticular, and particularly affects the large joints of the lower extremities, especially knees and ankles [11, 14, 15]. The main musculoskeletal imaging findings include joint effusion, synovial thickening and enhancement, tenosynovitis, enthesopathy and cellulitis (Figs. 4, 5).

Familial Mediterranean fever in an 8-year-old boy with knee pain. Sagittal T2-weighted MRI of the knee shows a moderate-size joint effusion (arrow). Arthritis in familial Mediterranean fever and tumor necrosis factor receptor-associated periodic syndrome (TRAPS) is usually nonerosive and monoarticular or oligoarticular. The absence of muscular edema and fasciitis on MRI helps in the differentiation from TRAPS

Familial Mediterranean fever in a 14-year-old boy with carpal pain. Transverse color Doppler sonogram shows thickening and hyperemia of the extensor digitorum tendon sheath (arrow), in keeping with tenosynovitis
Inflammation of the tunica vaginalis, which is an extension of the peritoneum, may lead to acute scrotal pain. Testicular ultrasound (US) may show unilateral epididymal and testicular enlargement, scrotal skin thickening and hydrocele. In addition, Doppler US is very helpful in the differential diagnosis with other causes of acute scrotum, particularly with testicular torsion, showing increased or normal testicular perfusion in children with acute scrotal pain [16] (Table 3).
Colchicine is the preferred treatment to prevent fever and systemic amyloidosis [17].
Tumor necrosis factor receptor-associated periodic syndrome
Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) is the second most frequent hereditary periodic fever disease. It has an autosomal dominant inheritance and is induced by mutations in the TNFRSF1A gene, which generates the tumor necrosis factor receptor [18]. TRAPS may present at any age, including in infants, and does not have a predilection for specific ethnicities [4].
Clinical presentation resembles that of familial Mediterranean fever and other periodic fever syndromes, systemic-onset juvenile idiopathic arthritis, Behçet disease, and PAPA syndrome, with febrile episodes, serositis, and articular and cutaneous manifestations. Distinctive features of TRAPS are conspicuous eye and skin symptoms, and longer attacks [18]. Polyserositis is more frequent but isolated pericarditis can occur, too (Fig. 6). Abdominal pain with tenderness resembling an acute abdomen may be due to peritonitis or inflammation of the abdominal wall musculature [19]. Ascites, retroperitoneal and mesenteric lymphadenopathy, chronic inflammation and fibrosis of the peritoneum, bowel obstruction due to adhesions, inflammation of the small bowel, and ulcerations in the duodenum, small intestine and colon have been reported in the literature (Fig. 7) [19, 20]. Articular involvement is generally monoarticular or oligoarticular, most commonly involving the knees, shoulders, elbows, hips, fingers, wrists and temporomandibular joints [21, 22]. Magnetic resonance imaging (MRI) may reveal underlying muscular edema, fasciitis and subcutaneous edema as increased T2 signal intensity within these areas [23, 24], representing the most distinctive imaging features of TRAPS.

Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) in a 5-year-old boy with fever and dyspnea. Posteroanterior chest radiograph shows cardiac enlargement with a “water-bottle” configuration (arrows) that corresponded to pericardial effusion on echocardiography (not shown)

Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) in an 11-year-old boy with fever, abdominal and musculoskeletal pain. Axial fat-suppressed T2-weighted (a) and axial T2-weighted MRI (b) of the abdomen show segments of small bowel wall thickening (straight arrows), mesenteric and retroperitoneal lymphadenopathy (arrowhead in b), and trace of free intraperitoneal fluid (curved arrow in a). Although clinical presentation mimicked acute appendicitis, the imaging differential diagnosis favored inflammatory bowel disease, Behçet disease and Henoch-Schönlein purpura, among others
The diagnosis of TRAPS is achieved with genetic testing [18]. Treatment depends on the severity of the manifestations. Children with occasional episodes may be treated with prednisone during the attacks, while in children with more frequent episodes, etanercept, an anti-tumor necrosis factor, may be effective in preventing the attacks. Anakinra (interleukin-1 receptor antagonist) and canakinumab (anti-interleukin-1 antibody) may also be beneficial in some children [4].
Hyperimmunoglobulin D syndrome/mevalonate kinase deficiency
Hyperimmunoglobulin D syndrome/mevalonate kinase deficiency results from autosomal recessive mutations in the MVK gene, which encodes mevalonate kinase, an important enzyme involved in the biosynthesis of nonsterol isoprenoid [25]. The mildest presentation of the disease known as hyperimmunoglobulin D syndrome is caused by decreased mevalonate kinase activity, while the more severe mevalonic aciduria is caused by lack of enzymatic activity [26].
Hyperimmunoglobulin D syndrome is defined by repeating febrile episodes, lymphadenopathy, hepatosplenomegaly, abdominal pain, and articular and cutaneous manifestation with onset in infancy [27]. Crises may be triggered by vaccination, infection, surgery, trauma and stress [28]. Some children present with a nondestructive arthritis, most frequently affecting the large joints and with a polyarticular distribution. Lymphadenopathy is more frequent in patients with hyperimmunoglobulin D syndrome than in familial Mediterranean fever [28].
Headache is the most frequent and often the only neurological symptom. However, some children present with developmental delay, ataxia, ocular symptoms and seizures, demonstrating the clinical continuum between mevalonic aciduria and hyperimmunoglobulin D syndrome [29]. MRI may demonstrate T2 hyperintensities in the white matter [30], cerebral atrophy, and more characteristically cerebellar atrophy (Fig. 8) [31, 32].

Hyperimmunoglobulin D syndrome in an 18-year-old girl who presented with headache and pyramidal signs. a Coronal fluid-attenuated inversion recovery (FLAIR) MRI of the brain shows a nonspecific subcortical focus of high signal intensity (arrow), a finding that has been described in patients with chronic headache in this entity. b Coronal T2-weighted MRI of the brain shows supra- and infratentorial parenchymal volume loss (arrows)
Although clinical findings, and serum IgD and urinary mevalonic aciduria analysis are useful for diagnosis, confirmation is established with genetic testing. Blockade of interleukin-1 with anakinra or canakinumab is useful in most patients [33].
Cryopyrin-associated periodic fever syndrome
Cryopyrin-associated periodic fever syndrome (CAPS) is a family of syndromes resulting from mutations in the C1AS1 gene, now known as NLRP3, on chromosome 1 [34]. Familial cold autoinflammatory syndrome and neonatal-onset multisystem inflammatory disease (NOMID) represent the two ends of a spectrum of disease activity. Children usually develop symptoms early in life with fever, cutaneous and articular manifestations.
Familial cold autoinflammatory syndrome
Familial cold autoinflammatory syndrome has an autosomal dominant inheritance and is typified by repeated brief febrile episodes, rash, polyarthralgia and conjunctivitis triggered by cold [35]. No specific imaging findings have been described in this entity.
Muckle–Wells syndrome
This is most commonly an autosomal dominant disorder, although sporadic cases may also happen. Recurrent febrile episodes with cutaneous, articular and ocular manifestations are characteristic. Fever is not always present [4, 36]. Joint involvement is usually in the form of oligoarthritis, predominantly affecting large joints. Sensorineural hearing loss is also common. Other neurological abnormalities are less common but multiple sclerosis-like lesions involving the periventricular area and the corpus callosum have been described on MRI in these patients [37]. Lymphadenopathy, abdominal pain and headache have been described as well (Fig. 9). Triggering factors are not commonly recognized.

Muckle-Wells syndrome (V200 M mutation in C1AS1 gene) in a 4-year-old girl with an 8-month history of rash, fatigue, decreased appetite and palpable enlarged lymph nodes. Axial contrast-enhanced CT images of the chest (a) and pelvis (b) show extensive bilateral axillary and iliac lymphadenopathy (arrows). Differential diagnosis includes other periodic inflammatory disorders, systemic-onset juvenile idiopathic arthritis, Behçet disease and lymphoproliferative disorders
Neonatal-onset multisystem inflammatory disease
Neonatal-onset multisystem inflammatory disease (NOMID), also called chronic infantile neurological cutaneous and articular syndrome, is associated with the most severe CAPS phenotype. Most cases of NOMID are sporadic, with only a minority having an autosomal dominant inheritance [38].
Children usually show cutaneous and articular manifestations, with fever at birth or in the first years of life [4]. However, fever may be absent. Osseous and articular inflammation may vary from arthralgia to severe arthropathy secondary to anomalous endochondral ossification and bony overgrowth [39]. Articular involvement is usually asymmetrical and mainly affects the knees. The initial imaging finding is physeal widening, with subsequent development of fraying and cupping of the metaphysis, which is irregular in outline, simulating rickets [40]. The characteristic radiographic finding is an unusual hypertrophy of the ossified part of the growth cartilage, appearing as a heterogeneous calcified mass with associated deformity of the adjacent epiphysis and metaphysis. The enlarged physis appears hypointense on T1- and T2-weighted images, and shows enhancement following intravenous contrast injection (Fig. 10) [40]. Decreased bone mineralization, conspicuous subperiosteal new bone formation along the diaphysis and metaphysis of the involved long bones, delayed bone age, genu varus or valgus, and leg length discrepancy may also be seen [4, 41, 42]. The differential diagnosis of the musculoskeletal findings of NOMID include rickets, chronic recurrent multifocal osteomyelitis, systemic-onset juvenile idiopathic arthritis and tuberculosis (Table 4).

Neonatal-onset multisystem inflammatory disease (NOMID) in a 13-year-old boy with recurrent fever, urticaria and right ankle and knee pain. a Lateral radiograph of the right knee at 8 years of age shows widening of the anterior aspect of the distal femoral physis with focal indentation of the adjacent epiphysis (arrowhead). b Lateral radiograph of the right knee 2 years later shows partial calcification of the enlarged distal femoral physis (arrow) and new widening of the anterior aspect of the proximal tibial physis by an unossified mass (arrowhead). c Anteroposterior radiograph 3 years later shows the progression of the ossification and physeal/epiphyseal enlargement at the distal femur and proximal tibia (straight arrows), and cupping of the distal femoral and proximal tibial metaphyses with sclerotic margin (curved arrows). d Sagittal T1-weighted MRI of the right knee shows hypointense enlargement of the distal femoral and proximal tibial physes (arrows) with associated deformity of the adjacent epiphyses (arrowheads) and prominent popliteal lymph nodes (asterisks)
Neurological manifestations are common. Computed tomography (CT) and MRI may show mild prominence of the ventricles and increased extra-axial fluid, suggestive of mild cerebral atrophy [36], as well as calcifications of the falx cerebri and dura mater, leptomeningeal enhancement and cochlear inflammation [4, 43].
Interleukin-1 targeted therapies appear to be useful including anakinra, rilonacept and canakinumab [44, 45].
Pyogenic arthritis pyoderma gangrenosum and acne syndrome
PAPA syndrome is an infrequent autoinflammatory disease inherited in an autosomal dominant pattern secondary to genetic alterations in the proline serine threonine phosphatase-interacting protein 1 (PSTPIP1/CD2BP1) gene, resulting in overproduction of interleukin-1β [4, 46].
PAPA syndrome usually presents early in life with arthritis, cystic acne, pyoderma gangrenosum and pathergy-like sterile abscesses at injection sites (Table 5). Arthritis is usually oligoarticular and destructive, mainly affecting the appendicular skeleton, including the elbow, knee, ankle and wrist (Fig. 11). Radiographic findings include osteophyte formation, generalized joint narrowing, subchondral sclerosis and cysts, periostitis and, less frequently, ankylosis [47, 48]. Osteolytic bone lesions, similar to those described in deficiency of interleukin-1 receptor antagonist and chronic recurrent multifocal osteomyelitis have also been described [49]. MRI findings are nonspecific, although they may show large joint effusions, synovial thickening and hyperenhancement, bone marrow edema and inflammatory changes in the soft tissues [47].

Pyogenic arthritis pyoderma gangrenosum and acne (PAPA) syndrome in a 2-year-old girl with slowly evolving right knee and ankle arthritis. a Sagittal contrast-enhanced fat-suppressed T1-weighted MRI of the right knee shows thickening and enhancement of the synovium (arrow) and reactive popliteal lymph node (arrowhead), in keeping with active synovitis. b Axial fat-suppressed T2-weighted MRI of the right ankle shows increased signal intensity in the tibiotalar joint that may represent effusion and/or synovial thickening (straight arrow), and tenosynovitis of the peroneal tendons (curved arrow)
Diagnosis at presentation may be tricky as clinical and imaging findings may mimic septic arthritis, osteomyelitis, juvenile idiopathic arthritis or chronic recurrent multifocal osteomyelitis (Table 6). In the setting of recurrent or chronic manifestations, other monogenic autoinflammatory disorders have to be included in the differential diagnosis. Therefore, clinical, laboratory and genetic findings should be put together to diagnose PAPA syndrome, which has to be considered in afebrile patients with history of recurrent joint inflammation, particularly after minor trauma, with purulent but aseptic synovial fluid [47]. Glucocorticoids are standard treatment, although etanercept, an anti-tumor necrosis factor and interleukin-1 antagonist, appears promising [50].
Interleukin-10 and interleukin-10 receptor deficiencies
Interleukin-10 and interleukin-10 receptor deficiencies are rare entities secondary to loss-of-function mutations in the genes encoding interleukin-10 or interleukin-10 receptor [50]. Clinical manifestations commonly begin early in life with inflammatory bowel disease, particularly with severe perianal involvement [51, 52] (Fig. 12). Other findings, such as folliculitis, recurrent respiratory disease, and arthritis, may also be present [53] (Table 5).

Interleukin-10 receptor mutation leading to interleukin-10 receptor alpha deficiency in a 13-month-old girl who presented with colitis, polyarthritis, rash and recurrent infections. a Transverse sonogram of the abdomen shows marked wall thickening of the sigmoid colon (arrows) with increased echogenicity of the surrounding fat (asterisk). b Coronal contrast-enhanced fat-suppressed T1-weighted MRI of the abdomen shows diffuse wall thickening and enhancement of the descending colon (arrow). c Longitudinal sonogram of the right hip shows moderate-size anechoic joint effusion (arrowhead and between calipers)
Interleukin-10 and interleukin-10 receptor deficiencies are characterized by a severe course with multifocal involvement, including abscesses, anal fissures and enterocutaneous and rectovaginal fistulae, usually requiring surgery [51]. Abdominal US may show bowel wall thickening and hyperemia with abnormal echogenicity and contours as well as increased echogenicity of the surrounding fat and abscess formation. Pelvic MRI is very useful in evaluating perianal disease as it allows precise delineation of fistulous tracts and identification of secondary fistulas or abscesses. The protocol used at our institution includes coronal short-TI inversion recovery and T1-weighted sequences, oblique axial fat-suppressed T2-weighted and diffusion, sagittal fat-suppressed T2-weighted, and oblique coronal and axial contrast-enhanced fat-suppressed T1-weighted sequences. Oblique axial and coronal images should be oriented orthogonal and parallel to the anal canal.
Remarkably, children show no response to immunosuppressive treatment, but allogeneic hematopoietic stem cell transplantation appears to lead to sustained remission [51, 54].
Adenosine deaminase 2 deficiency
Adenosine deaminase 2 deficiency is a recessive inherited autoinflammatory disease secondary to loss-of-function mutations in cat eye syndrome chromosome region 1 (CECR1), which generates an enzyme named adenosine deaminase type 2 involved in the maintenance of vascular endothelium and the development of monocytes [55, 56]. Affected patients can develop both vascular disease and immunodeficiency (Table 5).
Symptoms are mainly secondary to the involvement of medium- and small-sized arteries and vary greatly depending on the respective organ or system affected [56]. The most frequent presentation is early onset stroke, particularly before 5 years of age (Fig. 13), and the polyarteritis nodosa phenotype. In fact, adenosine deaminase 2 deficiency should be considered in children with stroke even if there is no evidence of systemic inflammation or cerebral vasculitis, as well as in patients with known polyarteritis nodosa and central nervous system involvement. Typically, stroke follows systemic inflammation and primarily involves lacunar infarctions and hemorrhages in the brainstem and deep brain nuclei [57, 58]. Unenhanced head CT is often the initial study in a child presenting with possible stroke and can rule out intracranial hemorrhage. However, CT has limited sensitivity for detecting acute ischemia and stroke mimics, such as complicated migraine syndromes, focal seizures, intracranial infections and intracranial neoplasms [59]. On CT, acute lacunar infarcts appear as small ill-defined hypodensities, while chronic lacunar infarcts show a similar density to cerebrospinal fluid. MRI is the preferred imaging technique for studying a suspected stroke. On MRI, acute lacunar infarcts appear slightly hypointense on T1-weighted images, hyperintense on fluid-attenuated inversion recovery (FLAIR) and T2-weighted images and show diffusion restriction on diffusion-weighted images. Chronic lesions are isointense to cerebrospinal fluid on all sequences but may demonstrate a peripheral hyperintense rim of marginal gliosis on FLAIR and T2-weighted images. In addition, in children with adenosine deaminase 2 deficiency, cerebral angiography does not reveal typical vasculitic findings [57].

Deficiency of adenosine deaminase 2 in a 5-year-old boy with an 1-week history of diplopia and unsteady gait. a Axial fluid-attenuated inversion recovery MRI of the brain shows a hyperintense focus in the right medial thalamus (arrow). b, c Axial diffusion-weighted (b) and axial apparent diffusion coefficient map (c) of the brain show diffusion restriction of the abnormality seen in (a) appearing as a hyperintense focus in (b) (arrow) and a hypointense focus in (c) (arrow), consistent with an acute arterial ischemic stroke. An old infarct is also noted in the left medial thalamus in (c) (arrowhead)
Anti-tumor necrosis factor therapy, adenosine deaminase 2-enzyme replacement therapy with fresh-frozen plasma or recombinant protein, or bone marrow transplant can be beneficial in the treatment of adenosine deaminase 2 deficiency [4, 55, 56, 60].
Pediatric sarcoidosis
Pediatric sarcoidosis is due to genetic alterations in the NOD2 gene and encompasses a group of pediatric granulomatous inflammatory disorders characterized by early atypical skin rash, recurrent uveitis and polyarthritis with marked synovial thickening and tenosynovitis [61,62,63] (Table 5). Blau syndrome and early-onset sarcoidosis are, respectively, the familial and sporadic forms, with the familial cases being inherited in an autosomal dominant pattern [64].
Non-caseating epithelioid giant cell granulomas are the distinctive pathological characteristic of pediatric sarcoidosis, likely representing an excessive immune-inflammatory reaction to a persistent antigen [64].
Arthritis is the most frequent manifestation and differential diagnosis with juvenile idiopathic arthritis can be challenging (Table 7). It usually develops in the first decade, often with minimal symptoms. It mostly presents as chronic nonerosive symmetrical polyarthritis affecting large and small peripheral joints, particularly wrists, knees, ankles and proximal interphalangeal joints [65]. Typically, tenosynovitis is more frequent and severe than intra-articular synovitis (Fig. 14) [66]. In fact, the flexor tendons of the digits and of the carpus, the extensor tendons of the ankle and the peroneal tendons may become very enlarged [64]. US usually reveals marked thickening of the tendon sheaths with increased vascularity on Doppler interrogation associated with synovial fluid [63]. In contrast, signs of intra-articular synovitis in the adjacent joint are not frequently present or are very mild.

Blau syndrome (familial form of pediatric sarcoidosis) in a 3-year-old girl with pain and swelling of the ankles for 2 months. a Axial fat-suppressed T2-weighted MRI of both ankles shows bilateral anterior tibial and peroneal tenosynovitis characterized by high T2 signal intensity fluid within the tendon sheaths (arrows). b Sagittal contrast-enhanced fat-suppressed T1-weighted MRI of the right ankle shows no evidence of articular synovitis. In children with pediatric sarcoidosis, tenosynovitis is much more frequent and severe than articular synovitis, being the best imaging clue in the differentiation from juvenile idiopathic arthritis. c Anteroposterior radiograph of the hand shows post-inflammatory changes including thin diaphysis of the second metacarpal bone (arrow), rotation and pseudo-collapse of the lunate and scaphoid (arrowheads), and ulnar shortening (asterisk)
Bone morphological changes may also be useful for diagnosis. They include camptodactyly, a contracture virtually limited to the proximal interphalangeal joints, carpal dysplasia with carpal crowding, loss of the normal distal radial epiphyseal anatomy with a biconcave articular surface, abnormal morphology of the distal ulna (plump ulna), short ulna, long thin diaphysis of the second metacarpal, and rotation and pseudo-collapse of the lunate and scaphoid (Fig. 13) [65].
Other clinical manifestations include interstitial pneumonitis, pericarditis, nephritis, small-vessel vasculitis, peripheral and mediastinal lymphadenopathy, cranial neuropathy and parotitis [61, 64]. Laboratory findings for pediatric sarcoidosis are nonspecific. Although elevated angiotensin converting enzyme levels may suggest sarcoidosis, the test is nonspecific in children and may be elevated in other systemic diseases. A definite diagnosis can be achieved with genetic testing. Systemic steroids, immunosuppressive and/or biological agents are the standard treatment for most children [62].
Conclusion
Imaging might provide clues for diagnosing autoinflammatory diseases but, given the wide variety and often nonspecific presentations of these diseases, its greatest value usually resides in delineating organ involvement and disease extent. Periodic fever syndromes have the articular involvement in common. However, familial Mediterranean fever and TRAPS may also cause peritonitis and pericardial and pleural effusions, while NOMID is characterized by an unusual hypertrophy of the ossified portion of the growth cartilage. PAPA syndrome is associated with a destructive arthritis. Interleukin-10 deficiency and interleukin-10 receptor deficiency syndrome should be considered in children with early-onset inflammatory bowel disease with severe perianal disease. Adenosine deaminase 2 deficiency should be considered in children with stroke and in children with known polyarteritis nodosa and central nervous system involvement. Severe tenosynovitis is the most characteristic feature of pediatric sarcoidosis.
References
McDermott MF, Aksentijevich I, Galon J et al (1999) Germline mutations in the extracellular domains of the 55 kda TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 97:133–144
Almeida de Jesus A, Goldbach-Mansky R (2013) Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol 147:155–174
Masters SL, Simon A, Aksentijevich I, Kastner DL (2009) Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 27:621–668
Barron KS, Kastner DL (2016) Periodic fever syndromes and other inherited autoinflammatory diseases. In: Petty RE, Laxer RM, Lindsley CB, Wedderburn LR (eds) Textbook of pediatric rheumatology, 7th edn. Elsevier, Philadelphia, pp 609–626
French FMF Consortium (1997) A candidate gene for familial Mediterranean fever. Nat Genet 17:25–31
Gershoni-Baruch R, Shinawi M, Leah K et al (2001) Familial Mediterranean fever: prevalence, penetrance and genetic drift. Eur J Hum Genet 9:634–637
Zissin R, Rathaus V, Gayer G et al (2003) CT findings in patients with familial Mediterranean fever during an acute abdominal attack. Br J Radiol 76:22–25
Aharoni D, Hiller N, Hadas-Halpern I (2000) Familial Mediterranean fever: abdominal imaging findings in 139 patients and review of the literature. Abdom Imaging 25:297–300
De Socio G, Cerquaglia C, Curigliano V et al (2009) Association between familial mediterranean fever and retroperitoneal fibrosis: retroperitoneal fibrosis regression after colchicine therapy. Int J Immunopathol Pharmacol 22:521–524
Gurkan OE, Dalgic B (2013) Gastrointestinal mucosal involvement without amyloidosis in children with familial Mediterranean fever. J Pediatr Gastroenterol Nutr 57:319–323
Ishak GE, Khoury NJ, Birjawi GA et al (2006) Imaging findings of familial Mediterranean fever. Clin Imaging 30:153–159
Kees S, Langevitz P, Zemer D et al (1997) Attacks of pericarditis as a manifestation of familial Mediterranean fever (FMF). QJM 90:643–647
Ince E, Cakar N, Tekin M et al (2002) Arthritis in children with familial Mediterranean fever. Rheumatol Int 21:213–217
Uthman I, Hajj-Ali RA, Arayssi T et al (2001) Arthritis in familial Mediterranean fever. Rheumatol Int 20:145–148
Brodey PA, Wolff SM (1975) Radiographic changes in the sacroiliac joints in familial Mediterranean fever. Radiology 114:331–333
Makay BB, Kefi A, Ünsal E (2007) Familial Mediterranean fever in the differential diagnosis of pediatric acute scrotum. Ege J Med 46:101–103
Gedalia A, Adar A, Gorodischer R (1992) Familial Mediterranean fever in children. J Rheumatol Suppl 35:1–9
Hull KM, Drewe E, Aksentijevich I et al (2002) The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 81:349–368
Chen YJ, Yu HH, Yang YH et al (2014) Recurrent abdominal pain as the presentation of tumor necrosis factor receptor-associated periodic syndrome (TRAPS) in an Asian girl: a case report and review of the literature. J Microbiol Immunol Infect 47:550–554
Alvarez-Lobos M, Hunter B, Cofré C et al (2006) Tumor necrosis factor receptor associated periodic syndrome (TRAPS). Report of two cases. Rev Med Chil 134:1558–1561
Stankovic K, Grateau G (2007) Auto inflammatory syndromes: diagnosis and treatment. Joint Bone Spine 74:544–550
Cantarini L, Lucherini OM, Cimaz R et al (2010) Sacroileitis and pericarditis: atypical presentation of tumor necrosis factor receptor-associated periodic syndrome and response to etanercept therapy. Clin Exp Rheumatol 28:290–291
Hull KM, Wong K, Wood GM et al (2002) Monocytic fasciitis: a newly recognized clinical feature of tumor necrosis factor receptor dysfunction. Arthritis Rheum 46:2189–2194
Dodé C, Papo T, Fieschi C (2000) A novel missense mutation (C30S) in the gene encoding tumor necrosis factor receptor 1 linked to autosomal-dominant recurrent fever with localized myositis in a French family. Arthritis Rheum 43:1535–1542
Goldstein JL, Brown MS (1990) Regulation of the mevalonate pathway. Nature 343:425–430
Drenth JP, Cuisset L, Grateau G et al (1999) Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International hyper-IgD study group. Nat Genet 22:178–1781
Mulders-Manders CM, Simon A (2015) Hyper-IgD syndrome/mevalonate kinase deficiency: what is new? Semin Immunopathol 37:371–376
van der Hilst JC, Bodar EJ, Barron KS et al (2008) Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore) 87:301–310
Haas D, Hoffmann GF (2006) Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis 1:13
Osborn RE, Alder DC, Mitchell CS (1991) MR imaging of the brain in patients with migraine headaches. AJNR Am J Neuroradiol 12:521–524
Ruiz Gomez A, Couce ML, Garcia-Villoria J et al (2012) Clinical, genetic, and therapeutic diversity in 2 patients with severe mevalonate kinase deficiency. Pediatrics 129:e535–e539
Bretón Martínez JR, Cánovas Martínez A, Casaña Pérez S et al (2007) Mevalonic aciduria: report of two cases. J Inherit Metab Dis 30:829
Galeotti C, Meinzer U, Quartier P et al (2012) Efficacy of interleukin-1-targeting drugs in mevalonate kinase deficiency. Rheumatology (Oxford) 51:1855–1859
Stahl N, Radin A, Mellis S (2009) Rilonacept--CAPS and beyond. Ann N Y Acad Sci 1182:124–134
Hoffman HM, Wanderer AA, Broide DH (2001) Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol 108:615–620
Neven B, Prieur AM, Quartier dit Maire P (2008) Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol 4:481–489
Compeyrot-Lacassagne S, Tran TA, Guillaume-Czitrom S et al (2009) Brain multiple sclerosis-like lesions in a patient with muckle-Wells syndrome. Rheumatology (Oxford) 48:1618–1619
Dávila-Seijo P, Hernández-Martín A, Torrelo A (2014) Autoinflammatory syndromes for the dermatologist. Clin Dermatol 32:488–501
Morbach H, Hedrich CM, Beer M, Girschick HJ (2013) Autoinflammatory bone disorders. Clin Immunol 147:185–196
Zaki FM, Sridharan R, Pei TS et al (2012) NOMID: the radiographic and MRI features and review of literature. J Radiol Case Rep 6:1–8
Torbiak RP, Dent PB, Cockshott WP (1989) NOMID – a neonatal syndrome of multisystem inflammation. Skeletal Radiol 18:359–364
Hill SC, Namde M, Dwyer A et al (2007) Arthropathy of neonatal onset multisystem inflammatory disease (NOMID/CINCA). Pediatr Radiol 37:145–152
Goldbach-Mansky R, Dailey NJ, Canna SW et al (2006) Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med 355:581–592
Lequerré T, Vittecoq O, Saugier-Veber P et al (2007) A cryopyrin-associated periodic syndrome with joint destruction. Rheumatology (Oxford) 46:709–714
Neven B, Marvillet I, Terrada C et al (2010) Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum 62:258–267
Smith EJ, Allantaz F, Bennett L et al (2010) Clinical, molecular, and genetic characteristics of PAPA syndrome: a review. Curr Genomics 11:519–527
Martinez-Rios C, Jariwala MP, Highmore K et al (2019) Imaging findings of sterile pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome: differential diagnosis and review of the literature. Pediatr Radiol 49:23–36
Lindor NM, Arsenault TM, Solomon H et al (1997) A new autosomal dominant disorder of pyogenic sterile arthritis, pyoderma gangrenosum, and acne: PAPA syndrome. Mayo Clin Proc 72:611–615
Caorsi R, Picco P, Buoncompagni A et al (2014) Osteolytic lesion in PAPA syndrome responding to anti-interleukin 1 treatment. J Rheumatol 41:2333–2334
Demidowich AP, Freeman AF, Kuhns DB et al (2012) Brief report: genotype, phenotype, and clinical course in five patients with PAPA syndrome (pyogenic sterile arthritis, pyoderma gangrenosum, and acne). Arthritis Rheum 64:2022–2027
Engelhardt KR, Shah N, Faizura-Yeop I et al (2013) Clinical outcome in IL-10- and IL-10 receptor-deficient patients with or without hematopoietic stem cell transplantation. J Allergy Clin Immunol 131:825–830
Glocker EO, Kotlarz D, Boztug K et al (2009) Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med 361:2033–2045
Kotlarz D, Beier R, Murugan D et al (2012) Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology 143:347–355
Kuşkonmaz B, Ayvaz D, Aydemir Y et al (2016) Successful outcome with second hematopoietic stem cell transplantation in a patient with IL-10R deficiency. Bone Marrow Transplant 51:615–616
Zhou Q, Yang D, Ombrello AK et al (2014) Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 370:911–920
Sahin S, Adrovic A, Barut K et al (2018) Clinical, imaging and genotypical features of three deceased and five surviving cases with ADA2 deficiency. Rheumatol Int 38:129–136
Elbracht M, Mull M, Wagner N et al (2017) Stroke as initial manifestation of adenosine deaminase 2 deficiency. Neuropediatrics 48:111–114
Bulut E, Erden A, Karadag O et al (2019) Deficiency of adenosine deaminase 2; special focus on central nervous system imaging. J Neuroradiol 46:193–198
Mirsky DM, Beslow LA, Amlie-Lefond C et al (2017) Pathways for neuroimaging of childhood stroke. Pediatr Neurol 69:11–23
Adler Y, Charron P, Imazio M et al (2015) ESC guidelines for the diagnosis and management of pericardial diseases. Rev Esp Cardiol (Engl Ed) 68:1126
Aróstegui JI, Arnal C, Merino R et al (2007) NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum 56:3805–3813
Rosé CD, Wouters CH, Meiorin S et al (2006) Pediatric granulomatous arthritis: an international registry. Arthritis Rheum 54:3337–3344
Ikeda K, Kambe N, Satoh T et al (2013) Preferentially inflamed tendon sheaths in the swollen but not tender joints in a 5-year-old boy with Blau syndrome. J Pediatr 163:1525.e1
Rosé CD, Wouters C (2016) Pediatric sarcoidosis. In: Petty RE, Laxer RM, Lindsley CB, Wedderburn LR (eds) Textbook of pediatric rheumatology, 7th edn. Elsevier, Philadelphia, pp 517–525
Rosé CD, Pans S, Casteels I et al (2015) Blau syndrome: cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatology (Oxford) 54:1008–1016
Ikeda K, Kambe N, Takei S et al (2014) Ultrasonographic assessment reveals detailed distribution of synovial inflammation in Blau syndrome. Arthritis Res Ther 16:R89
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Navallas, M., Inarejos Clemente, E.J., Iglesias, E. et al. Autoinflammatory diseases in childhood, part 1: monogenic syndromes. Pediatr Radiol 50, 415–430 (2020). https://doi.org/10.1007/s00247-019-04536-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-019-04536-9