
Abstract
Background
Because of its absence of ionising radiation and possibility for obtaining functional information, MRI is promising for assessing lung disease in children who require repetitive imaging for long-term follow-up.
Objective
To describe MRI findings in children with cystic fibrosis and evaluate semi-quantitative dynamic contrast-enhanced lung perfusion.
Materials and methods
We retrospectively compared lung MRI in 25 children and young adults with cystic fibrosis (median age 3.7 years) to 12 children (median age 2 years) imaged for other pathologies. MRI at 1.5 T included respiratory-gated sequences and contrast-enhanced lung perfusion imaging. We described and graded any morphologic change. Signal enhancement and time to peak values of perfusion abnormalities were compared to those of normally enhancing lung parenchyma.
Results
Frequent findings in patients with cystic fibrosis were bronchial wall thickening (24/25, 96%), areas of consolidation (22/25, 88%), enlarged lymph nodes (20/25, 80%), bronchiectasis (5/25, 20%) and mucus plugging (3/25, 12%). Compared to normally enhancing lung, perfusion defects (21/25, 84%), characterised by decreased enhancement, showed prolonged time to peak. Areas of consolidation showed increased enhancement. While time to peak of procedure-related atelectasis was not significantly different from that of normal lung, disease-related consolidation showed prolonged time to peak (P=0.01).
Conclusion
Lung MRI demonstrates structural and perfusion abnormalities in children and young people with cystic fibrosis. Semi-quantitative assessment of dynamic contrast-enhanced perfusion imaging might allow differentiation between procedure-related atelectasis and disease-related consolidation.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In cystic fibrosis, the most common genetic disease affecting the European population with an incidence of 1:2,000–3,500 live births, the main cause of morbidity and mortality is still lung disease, which limits life expectancy [1, 2]. The high viscous secretions lead to airway obstruction, bacterial infection, bronchiectasis and progressive destruction of the lung parenchyma [3]. The pulmonary manifestations of cystic fibrosis begin early in childhood and require continuous medical care, including repeated imaging of the chest for surveillance of the lung changes and monitoring of the medical therapy. Standard imaging protocols in children and young adults with cystic fibrosis include regular chest radiography or high-resolution CT [4, 5]. However radiographs are limited in their depiction of early parenchymal abnormalities, and both CT and radiographs involve ionising radiation, which can accumulate to substantial effective doses when applied repeatedly from early childhood into adulthood and therefore should be avoided in small children. At some centres, MRI of the lung has been introduced for monitoring people with cystic fibrosis, without radiation exposure [6, 7]. Despite technical challenges for imaging lung parenchyma, such as susceptibility artefacts, low proton density and signal loss from respiratory motion, several MRI techniques have been proposed for assessing lung structure, with results comparable to those of CT [8, 9]. Furthermore MRI has the potential for delivering additional functional information [10, 11]. Based on recommendations from the literature [7, 12,13,14], we have implemented a clinical MRI protocol for imaging lung structure and perfusion at our department [15]. In this paper we describe MRI findings in children and young adults with cystic fibrosis in comparison to those without cystic fibrosis after semi-quantitatively evaluating dynamic contrast-enhanced lung perfusion of perfusion defects and consolidations in comparison to normal lung tissue.
Materials and methods
Patients
The MRI studies of 37 consecutive patients (21 [57%] male, 16 [43%] female), referred for MRI assessment of the lungs between December 2012 and December 2013, were retrospectively evaluated for morphologic and perfusion abnormalities. According to Swiss legislation at the time the study was initiated, retrospective analysis of our own data was exempt from formal research ethics board approval and informed consent was waived. The study was reported to the government as required by law. As part of the clinical hospital routine, patients and/or parents had consented to MRI with contrast administration and sedation when applicable. The MRI findings in 25 patients with stable cystic fibrosis (median age 3.7 years, interquartile range 1.4–6.4 years, range 1.0–19.9 years), imaged for monitoring lung involvement, were compared to those of 12 children (median age 2.0 years, interquartile range 0.6–5.5 years, range 0.3–9.4 years) imaged for pathologies other than cystic fibrosis (diagnoses and patient characteristics are given in Table 1).
Magnetic resonance imaging
All MRI examinations were performed on a 1.5-T system (Discovery MR 450; GE Healthcare, Waukesha, WI) using a 32-channel cardiac coil (n=27) or smaller flexible multichannel surface coil (n=10) depending on the size of the child. Most of the children were imaged under sedation (n=30) with propofol (Propofol-Lipuro 1%; Braun Medical AG, Sempach, Switzerland) applied by anaesthesiologists according to hospital routine. The imaging protocol included non-gated steady-state free precession gradient-echo sequences (FIESTA) in three orthogonal planes, axial and coronal single-shot fast spin-echo sequences, axial respiratory-gated T2-weighted fast spin-echo sequence with periodically rotated overlapping parallel lines with enhanced reconstruction (PROPELLER), coronal respiratory-gated proton-density-weighted fast spin-echo sequence, as well as axial and coronal T1-weighted fast spin-echo or fast spoiled gradient-echo acquisitions before and after intravenous contrast injection. The technical details are given in Table 2. Perfusion imaging was performed with a dynamic 3-D gradient-echo sequence (TRICKS). By under-sampling of the peripheral k-space and parallel imaging, we achieved a temporal resolution of 1–2 s. Image acquisition was started together with manual intravenous injection of a short bolus of gadolinium-based contrast agent (0.1 mmol/kg bodyweight, gadoteric acid, Dotarem, Guerbet AG, Zürich, Switzerland; or gadodiamide, Omniscan, GE Healthcare AG, Opfikon, Switzerland) and comprised 40 phases (duration 40–80 s). Total imaging time including time for sedation ranged 30–40 min; mean sequence acquisition time was 20 min.
Image analysis
All MRI studies were reviewed by two radiologists (C.J.K. and C.A., 18 years and 1 year of experience in paediatric radiology) in a consensus reading on the department’s picture archiving and communication system (PACS, IDS7; Sectra Medical Systems, Linköping, Sweden) and by another radiologist (R.G., 1 year experience in paediatric radiology) separately for assessing inter-reader agreement for structural and perfusion findings. For morphologic assessment, we viewed all MRI sequences but relied mainly on the respiratory-gated fast spin-echo sequences. Although any morphologic abnormality was noted, we focused the evaluation on findings expected in cystic fibrosis lung disease. We modified the scoring system described by Eichinger et al. [16] in order to separately assess bronchial wall thickening and bronchiectasis, to differentiate between procedure-related and disease-related consolidations, and to assess lymphadenopathy.
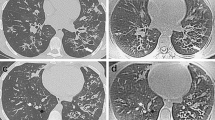
We assessed and graded the following items: (1) Bronchial wall thickening was defined as visible double lines in the central lung zone (at segmental and subsegmental levels) with higher signal intensity than that of other interstitial lung markings (Figs. 1 and 2). (2) Bronchiectasis was defined as visible bronchi within 1 cm of the pleural surface in the peripheral lung zone or as abnormal widened bronchial tubes with loss of tapering in the central long zones (Figs. 1 and 2). (3) Mucus plugging was defined as high signal intensity seen within a dilated bronchus (Figs. 1 and 2). These three items were graded on a 4-point scale based on the number of involved lung segments: absent (0 segments), mild (1–5 segments), moderate (6–9 segments) and severe (>9 segments).

Axial respiratory-gated fat-saturated T2-weighted fast spin-echo MR images with radial readout (PROPELLER, TR/TE >1,800/52–80 ms) show morphologic findings in children with cystic fibrosis. a Bronchial wall thickening is shown as double lines (arrows) with high signal intensity in a 13-year-old boy. b Bronchiectasis is shown as visible bronchi (arrows) within 1 cm of the pleural surface and loss of tapering in a 16-year-old girl. c Mucus plugging is evident as high signal intensity obliterating the lumen of dilated bronchi (arrows) in a 6-year old girl. d Lymphadenopathy is shown as mildly enlarged tracheobronchial and mediastinal nodes (arrows) in a 6-year-old girl. TE echo time, TR repetition time

Axial respiratory-gated fat-saturated T2-weighted fast spin-echo MR image with radial readout (PROPELLER, TR/TE 5,700/55 ms) in a 6-year-old girl with cystic fibrosis shows dilated bronchi of variable degree in both upper lobes, with bronchial wall thickening (arrows) and mucus plugging (arrowheads). Note eccentric wall thickening in severely dilated bronchus (*). TE echo time, TR repetition time
Areas of consolidation larger than 1 cm, with homogeneous high signal intensity on morphologic images (Figs. 3 and 4), with or without presence of an air bronchogram, were divided into procedure-related atelectasis and disease-related consolidations based on morphologic criteria: (4) Procedure-related atelectasis (Fig. 3) was diagnosed when the lesion was not visible at the beginning of the examination, developed and enlarged during the examination, was located in posterior dependent lung portions and showed irregular margins. (5) Disease-related consolidation (Fig. 4) was defined as wedge-shaped area with straight and sharp borders, visible from the beginning of the examination, without change during the study and usually located in non-dependent ventral or lateral portions of the lung. The extent of consolidations was graded on a 4-point scale based on the number of involved lung lobes: absent (0 lobes), mild (1 lobe), moderate (2 lobes), severe (3 or more lobes). (6) Lymphadenopathy was graded based on the involvement of the hila and mediastinum: absent (no enlarged nodes), mild (1 site), moderate (2 sites), severe (3 sites). Based on normative data for CT [17], we defined lymph nodes as enlarged when their short-axis diameter exceeded 4 mm for tracheobronchial nodes and 6 mm for mediastinal nodes (Fig. 1).

Axial images show procedure-related atelectasis in a 3-year-old boy with cystic fibrosis. a Initially acquired steady-state free precession image (FIESTA, TR/TE 3/1 ms, flip angle 45°) shows streaky subpleural infiltrates (arrows in a), which progress to consolidations (arrows in b) with homogeneous high signal intensity and irregular borders on contrast-enhanced fat-saturated fast spoiled gradient-echo image (FSPGR, TR/TE 140/1.3 ms, flip angle 60°) obtained at the end of the examination (b). TE echo time, TR repetition time

Axial images show disease-related consolidation (arrows) in a 6-year-old girl with cystic fibrosis. A wedge-shaped and sharply delineated high signal intensity area is shown already on the steady-state free precession image (FIESTA, TR/TE 3/1 ms, flip angle 45°) acquired at the beginning of the study (a), which is unchanged on the T1-weighted contrast-enhanced fast spin-echo image (FSE, TR/TE 550/11 ms) obtained at the end of the examination (b). TE echo time, TR repetition time
We performed qualitative assessment of lung perfusion on parametric images (subtraction images at signal peak enhancement of the lungs, and enhancement integral representing pulmonary blood volume) (Fig. 5). Perfusion defects were defined as areas with lower signal intensity on subtraction images and lower blood volumes on perfusion maps than that of the normal surrounding lung tissue. The extent of perfusion defects was graded similarly to consolidations based on the number of involved lung lobes. Semi-quantitative evaluation of lung perfusion was performed by one radiologist (C.A.) on a commercially available workstation (VolumeShare 5 Advantage Workstation AW 4.6; GE Healthcare, Waukesha, WI). After constructing respective parametric maps from the 3-D dynamic contrast-enhanced series, we measured signal enhancement and time to peak values in normal lung, perfusion defects and consolidations using a region of interest (ROI) with a fixed diameter (2 cm).

Perfusion images in a 15-year-old girl with cystic fibrosis. a Perfusion map (signal enhancement integral) and (b) subtraction image at peak enhancement illustrate perfusion defects (arrows)
One reader (C.A.) assessed overall image quality of the respiratory-gated fast spin-echo sequences for delineation of structural lung changes on a 5-point Likert scale: score 1 = excellent; score 2 = good; score 3 = mild impairment; score 4 = moderate impairment, still interpretable; score 5 = severe impairment, nondiagnostic.
Statistical analysis
Descriptive data are given as mean ± standard deviation for continuous variables with normal distribution and as median (interquartile range) for variables without normal distribution. We used the chi-squared test to compare the frequencies of morphologic characteristics between the groups with and without cystic fibrosis. We compared the graded morphologic and qualitative perfusion data in mild, moderate or severe grades using the Mann–Whitney test. We compared semi-quantitative perfusion data among different abnormalities with independent samples t-test and to ipsilateral normal lung parenchyma with paired samples t-test. A two-tailed P-value below 0.05 was considered to indicate statistical significance. We assessed accuracy of peak enhancement delay measurements for differentiating between procedure-related atelectasis and disease-related consolidation using receiver operating characteristic (ROC) analysis. Un-weighted Cohen kappa statistic was used to assess inter-reader agreement of morphologic and qualitative perfusion findings (κ = 1–0.81, excellent agreement; κ = 0.8–0.61, good agreement; κ = 0.6–0.41, moderate agreement; κ=0.4–0.21, fair agreement; κ=0.2–0, poor agreement). All statistical analyses were performed using commercially available software packages (IBM SPSS Statistics, release 22, IBM, Armonk, NY; or MedCalc Statistical Software version 16.2.0, MedCalc Software, Ostend, Belgium).
Results
Image quality
All MRI studies were obtained without complication and were considered diagnostically sufficient by the original readers. None of the patients with cystic fibrosis underwent additional CT, and 1/12 (8%) patients without cystic fibrosis required additional high-resolution CT looking for interstitial lung disease. Overall image quality of the respiratory-gated fast spin-echo sequences was excellent in 13 (35%), good in 19 (51%), mildly impaired in 4 (11%) and moderately impaired in 1 (3%) of the 37 MRI studies. No study was considered nondiagnostic.
Morphology
The frequency and grades of structural pulmonary findings in all children with cystic fibrosis younger than 5 years and in those without cystic fibrosis are given in Table 3. Bronchial wall thickening occurred in all but one patient with cystic fibrosis and was significantly more frequent in this group, with involvement of more lung segments in the cystic fibrosis group than in the no cystic fibrosis group. Whilst bronchiectasis was only seen in patients with cystic fibrosis (5/25, 20%), mucus plugging was rare but found in both the cystic fibrosis and no cystic fibrosis groups (3/25, 12% versus 1/12, 8%). On the other hand, areas of consolidation were observed with high frequency in both groups (22/25, 88% versus 9/12, 75%), mainly from an overall high rate of atelectasis (22/37, 59%) from sedation. However disease-related consolidation was seen in a third of the patients with cystic fibrosis (8/25, 32%) but only in one patient without cystic fibrosis (1/25, 8%). Enlarged hilar and mediastinal lymph nodes were significantly more frequent in people with cystic fibrosis (20/25, 80% versus 2/12, 17%) and often involved several sites. Small amounts of pleural fluid were visible in 19 cases, with a similar frequency in both groups.
Other pulmonary findings were six hyperinflated or cystic lung areas in four patients with congenital lung malformations, and these were evident on respiratory-gated fast spin-echo sequences as low signal intensity areas without any internal lung structures or vessels.
Lung perfusion
The children and young adults with cystic fibrosis more often showed perfusion defects than children without cystic fibrosis (21/25, 84% versus 4/12, 33%), with involvement of more lung lobes (Table 3). Whereas in patients with cystic fibrosis perfusion defects were seen in areas of structurally normal-appearing lung, perfusion defects in the no cystic fibrosis group were restricted to the cystic or focally hyperinflated lung areas and were not considered for quantitative analysis.
Semi-quantitative perfusion data of 25 perfusion defects, 26 areas of consolidation (17 procedure-related atelectasis and 9 disease-related consolidations) and ipsilateral normal lung parenchyma are illustrated in Figs. 6 and 7. Comparison of perfusion data in perfusion abnormalities to those of ipsilateral normal lung parenchyma is given in Tables 4 and 5. Perfusion defects, characterised by decreased signal enhancement, showed significantly longer time to peak values than normal lung (14.2±3.7 s, 95% confidence interval 12.7–15.8 s, versus 10.9±2.6 s, 95% confidence interval 10.0–11.8 s; P=0.0001, independent samples t-test). All areas of consolidation demonstrated significantly higher signal enhancement than normal lung (1,235±503, 95% confidence interval 1,032–1,438, versus 390±139, 95% confidence interval 343–437; P<0.0001, independent samples t-test), but the degree of signal enhancement was not significantly different between procedure-related atelectasis and disease-related consolidations (1,330±522, 95% confidence interval 1,062–1,599, versus 1,055±437, 95% confidence interval 720–1,391; P=0.19, independent samples t-test). Disease-related consolidations showed significantly longer time to peak values (13.5±2.9 s, 95% confidence interval 11.3–15.7 s) than normal lung (P=0.01, independent samples t-test), not significantly different from those of perfusion defects (14.2±3.7 s, 95% confidence interval 12.7–15.8 s, P=0.6, independent samples t-test). Time to peak values of procedure-related atelectasis (10.9±2.1 s, 95% confidence interval 9.8–12.0 s) was not significantly different from those of all normal lung measurements (independent samples t-test, P=0.95). Comparing procedure-related atelectasis to ipsilateral normal lung parenchyma in the same patient, the time to peak was slightly prolonged by 0.5±0.8 s (paired samples t-test, P=0.02; Table 5). Disease-related consolidations showed a 2.6±1.0 s longer time to peak value than atelectasis (independent samples t-test, P=0.01).

Box-and-whisker plots of signal enhancement integral obtained by contrast-enhanced perfusion MRI in normal lung parenchyma, perfusion defects, procedure-related atelectasis and disease-related consolidation. Central boxes represent values from the lower to upper quartiles. Middle lines represent the median. Lines at the end of the whiskers represent the minimum and maximum values

Box-and-whisker plots of time to peak values obtained by contrast-enhanced perfusion MRI in normal lung parenchyma, perfusion defects, procedure-related atelectasis and disease-related consolidation. Central boxes represent values from the lower to upper quartile. Middle lines represent the median. Lines at the end of the whiskers represent the minimum and maximum values, excluding outliers (defined as a value that is smaller than the lower quartile minus 1.5 times the interquartile range, or larger than the upper quartile plus 1.5 times the interquartile range), which are shown as data points outside the whiskers (circles)
The ROC analysis of the peak enhancement delays in areas of consolidation indicated a sensitivity of 94% and specificity of 67% for recognising procedure-related atelectasis by a cutoff value of ≤1.6 s as determined by the Youden J index. The area under the curve was 0.856 (95% confidence interval 0.685–1.000; P<0.0001; Fig. 8).

Receiver operating characteristic curve for peak enhancement delay. Area under the curve 0.856 (95% confidence interval 0.685–1.000, P<0.0001). Circle represents the optimal cut-off value of ≤1.6 s (Youden J index) for differentiating procedure-related atelectasis from disease-related consolidation, with a sensitivity of 94% and specificity of 67%
Inter-reader agreement
Inter-reader statistics are listed in Table 6. Assessing the presence and grading the extent of bronchial wall thickening, bronchiectasis, areas of consolidation, lymphadenopathy and perfusion defects showed good to excellent agreement. Only the assessment of mucus plugging showed fair agreement.
Discussion
With this study, we confirmed that current MRI techniques can show structural lung findings in children, emphasising a minor opinion in current literature that MRI is feasible for imaging early cystic fibrosis [11], which is in contrast to the widespread belief that MRI is not suited for assessing young children [18]. The expected morphologic lung findings, such as bronchial wall thickening, bronchiectasis and consolidation, were demonstrated in children and young adults with cystic fibrosis with a much higher frequency than in the children with other pathologies. The detection of bronchial wall thickening by MRI in our patients with cystic fibrosis (24/25, 96%) was significantly higher than in children without cystic fibrosis (3/12, 25%) and also higher than reported for CT in infants with cystic fibrosis (45%) [19]. We assume that MRI, where not just thickness of the bronchial wall but also increased signal intensity might contribute to detection of bronchial wall thickening, could be more sensitive than CT for detecting airway inflammation. It has been speculated that oedema, increased cellularity and hyperaemia of the bronchial wall and surrounding tissues could be a reasonable explanation for the high detection rate of “bronchial wall thickening” on T2-weighted and contrast-enhanced T1-weighted images [11]. However further studies with clinical correlation are needed to investigate whether bronchial wall thickening seen on CT or MRI is the better indicator for airway inflammation. In contrast to previous MRI studies, where bronchial wall thickening and bronchiectasis were assessed together on single-shot sequences [16], we were able to separately assess these two items because of the higher spatial and contrast resolution of our respiratory-gated T2-weighted fast spin-echo sequences with radial k-space readout (PROPELLER) [15].
In our study, the frequency of detecting bronchiectasis in young children with cystic fibrosis (1/17, 12%, <5 years of age) by MRI was much lower than that reported for CT in infants and young children with cystic fibrosis (up to 61.5% at 3 years of age) [20], indicating that mild bronchiectasis without inflammation is potentially missed by MRI. However the different study populations might also account for the difference in the frequency of detected bronchiectasis. Although MRI has been shown to accurately assess the extent and severity of advanced bronchiectasis in older patients [21], improved MRI techniques are still needed for depicting mild dilatation of peripheral bronchi.
This shortcoming might be outweighed by the unique ability of MRI to assess perfusion abnormalities in the peripheral lung, a sequelae of small airway disease already present in early cystic fibrosis lung disease even without discernible bronchiectasis [8, 11]. Dynamic contrast-enhanced MRI shows impaired lung ventilation indirectly by perfusion deficits from hypoxic vasoconstriction [12, 22], similarly to lung perfusion scintigraphy but without radiation exposure [23,24,25]. In our young cystic fibrosis group, 14/17 (82%) children showed perfusion defects with mild (5/14, 36%), moderate (6/14, 43%) or severe (3/14, 21%) grades in lung areas with normal-appearing parenchyma. In the control group, there were significantly fewer perfusion defects (4/12, 33%), which were only seen in focal areas of hyperinflation or cysts but not in normal-appearing lung parenchyma. Therefore, our results confirm that perfusion deficits in morphologically normal-appearing lung parenchyma appear to be typical for small airway disease, which is a recognised feature of early cystic fibrosis [11].
Mucus plugging was a rare finding overall (4/37 patients, 11%) without significant difference between both groups because there was one infant in the no cystic fibrosis group with complete atelectasis of the right lung from secretion-filled bronchi. The low detection rate of mucus plugging in our patients with cystic fibrosis (3/25, 12%) is in contrast to the results from Wielpütz et al. [11], who found mucus plugging to be the second most frequent structural MRI finding in infants and preschool children, with a high rate of 63%. Although this could be from differences in patient populations or clinical status, we speculate that the higher spatial and contrast resolution of our respiratory-gated T2-weighted sequence enabled us to better discriminate between bronchial wall thickening and mucus plugging, thus avoiding over-diagnosis of the latter.
Conversely, parenchymal consolidation (including procedure-related atelectasis and disease-related consolidation) was a more frequent finding in our patients with cystic fibrosis (22/25, 88%), mainly because of the high rate of procedure-related atelectasis (14/25, 56%) also present in the no cystic fibrosis group (8/12, 67%). Patchy consolidation in dependent posterior lung areas is a common problem in cross-sectional lung imaging under anaesthesia [26] and was accentuated in our study by sedation with propofol without the use of controlled ventilation. Nonetheless we were able to distinguish disease-related consolidation from procedure-related atelectasis with regard to location, shape and the time course of the consolidation. By considering anteriorly or laterally located lung areas of high signal intensity with straight borders and being present throughout the MRI examination as disease-related consolidation, we found more disease-related consolidations in patients with cystic fibrosis (8/25, 32%) than in patients without cystic fibrosis (1/12, 8%). In addition, our results show that semi-quantitative perfusion analysis might help in the discrimination between atelectasis from sedation and consolidation as expression of cystic fibrosis lung disease. While procedure-related atelectasis showed similar peak enhancement to normal lung, disease-related consolidation showed delayed peak enhancement (around 3 s later). With the contrast travel time between pulmonary artery and aorta being in the range of 4–5 s [27], delayed peak enhancement of consolidations indicates predominant systemic blood supply from the bronchial arterial system.
To our knowledge this is the first attempt to semi-quantitatively characterise lung perfusion abnormalities by dynamic contrast-enhanced MRI in children. Perfusion defects had been assessed only qualitatively by the signal intensity of the lung parenchyma at signal peak enhancement [28, 29]. With the assessment of time to peak and signal enhancement integral during the first pass of contrast (as estimate of the blood volume) on parametric maps, and comparing respective measurements to those of normal lung parenchyma, we could specify that perfusion defects seen in cystic fibrosis demonstrate half of the normal blood volume and delayed perfusion by 3 s on average. Besides assessing the extent of perfusion defects, such quantitative measures of perfusion might open a new possibility to grade the severity of small airway disease.
Another feature worth considering for grading cystic fibrosis lung disease is lymphadenopathy as an estimate of inflammatory activity. In our patients with cystic fibrosis we noted a higher frequency (20/25, 80%) of mildly enlarged hilar and mediastinal lymph nodes than in patients without cystic fibrosis (2/12, 17%, P=0.002, chi-squared test). The good to excellent inter-reader agreement for grading the majority of the assessed morphologic and perfusion abnormalities by relatively inexperienced readers in our study was in the same range as reported for CT-scoring systems [30], which confirms the utility of MRI for assessing the severity and monitoring cystic fibrosis lung disease [4]. Only the assessment of mucus plugging showed fair agreement, which can be explained by the low rate in our patients and underscores the aforementioned difficulties in differentiating between bronchial wall thickening and mucus plugging in peripheral small airways by current MRI techniques [4].
Major limitations of our study are its retrospective nature, lack of direct comparison with other imaging modalities and lack of a control group of healthy children. Although comparison of grading cystic fibrosis lung disease by MRI, CT and ventilation scintigraphy would be desirable to assess the performance of MRI, such a study involving significant radiation exposure appears unethical since the feasibility of MRI has been established [4]. Because subjecting healthy children to MRI under sedation and application of gadolinium-based contrast agent for research purposes would also be unethical, the only possible control group for this study was children without cystic fibrosis undergoing chest MRI that included the sequences required for assessing the lungs. Based on our results alone, we cannot recommend that a perfusion study be routinely included in MRI for assessing cystic fibrosis lung disease. However other groups have also shown that perfusion analysis can be valuable for grading cystic fibrosis lung disease by MRI [11, 31]. Further prospective studies with larger cohorts are required to determine structural and perfusion lung MRI findings in cystic fibrosis patients with different severity of lung disease and to assess their relevance for introducing and monitoring medical therapy.
Conclusion
Current lung MRI techniques can demonstrate the typical structural findings of cystic fibrosis lung disease, even in young children. Semi-quantitative assessment of dynamic contrast-enhanced lung perfusion imaging can further characterise perfusion abnormalities, which is valuable for differentiating between disease-related consolidation and sedation-related atelectasis and could potentially be used for grading the severity of lung perfusion defects seen in early cystic fibrosis lung disease. Therefore MRI may be considered as an advanced imaging modality in the routine surveillance of cystic fibrosis lung disease to avoid the radiation exposure inherent to CT and scintigraphy.
References
Gibson RL, Burns JL, Ramsey BW (2003) Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168:918–951
Moskowitz SM, Gibson RL, Effmann EL (2005) Cystic fibrosis lung disease: genetic influences, microbial interactions, and radiological assessment. Pediatr Radiol 35:739–757
Stoltz DA, Meyerholz DK, Welsh MJ (2015) Origins of cystic fibrosis lung disease. N Engl J Med 372:351–362
Wielpütz MO, Eichinger M, Biederer J et al (2016) Imaging of cystic fibrosis lung disease and clinical interpretation. Rofo 188:834–845
Kerem E, Conway S, Elborn S et al (2005) Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros 4:7–26
Henzler T, Schmid-Bindert G, Schoenberg SO et al (2010) Diffusion and perfusion MRI of the lung and mediastinum. Eur J Radiol 76:329–336
Puderbach M, Eichinger M (2010) The role of advanced imaging techniques in cystic fibrosis follow-up: is there a place for MRI? Pediatr Radiol 40:844–849
Biederer J, Mirsadraee S, Beer M et al (2012) MRI of the lung (3/3)-current applications and future perspectives. Insights Imaging 3:373–386
Wielpütz M, Kauczor HU (2012) MRI of the lung: state of the art. Diagn Interv Radiol 18:344–353
Eichinger M, Tetzlaff R, Puderbach M et al (2007) Proton magnetic resonance imaging for assessment of lung function and respiratory dynamics. Eur J Radiol 64:329–334
Wielpütz MO, Puderbach M, Kopp-Schneider A et al (2014) Magnetic resonance imaging detects changes in structure and perfusion, and response to therapy in early cystic fibrosis lung disease. Am J Respir Crit Care Med 189:956–965
Biederer J, Heussel CP, Puderbach M et al (2014) Functional magnetic resonance imaging of the lung. Semin Respir Crit Care Med 35:74–82
Eichinger M, Puderbach M, Fink C et al (2006) Contrast-enhanced 3D MRI of lung perfusion in children with cystic fibrosis - initial results. Eur Radiol 16:2147–2152
Wild JM, Marshall H, Bock M et al (2012) MRI of the lung (1/3): methods. Insights Imaging 3:345–353
Gnannt R, Amaxopoulou CE, Goetti R et al (2014) Qualitative assessment of MRI sequences for morphological imaging of the lungs in children with cystic fibrosis. Pediatr Radiol 44:308
Eichinger M, Optazaite DE, Kopp-Schneider A et al (2012) Morphologic and functional scoring of cystic fibrosis lung disease using MRI. Eur J Radiol 81:1321–1329
de Jong PA, Nievelstein RJ (2012) Normal mediastinal and hilar lymph nodes in children on multi-detector row chest computed tomography. Eur Radiol 22:318–321
Rossi UG, Owens CM (2005) The radiology of chronic lung disease in children. Arch Dis Child 90:601–607
Sly PD, Brennan S, Gangell C et al (2009) Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med 180:146–152
Sly PD, Gangell CL, Chen L et al (2013) Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med 368:1963–1970
Puderbach M, Eichinger M, Haeselbarth J et al (2007) Assessment of morphological MRI for pulmonary changes in cystic fibrosis (CF) patients: comparison to thin-section CT and chest x-ray. Investig Radiol 42:715–725
Swift AJ, Telfer A, Rajaram S et al (2014) Dynamic contrast-enhanced magnetic resonance imaging in patients with pulmonary arterial hypertension. Pulm Circ 4:61–70
Mirsadraee S, van Beek EJ (2015) Functional imaging: computed tomography and MRI. Clin Chest Med 36:349–363
Altes TA, Eichinger M, Puderbach M (2007) Magnetic resonance imaging of the lung in cystic fibrosis. Proc Am Thorac Soc 4:321–327
Rajaram S, Swift AJ, Telfer A et al (2013) 3D contrast-enhanced lung perfusion MRI is an effective screening tool for chronic thromboembolic pulmonary hypertension: results from the ASPIRE registry. Thorax 68:677–678
Newman B, Krane EJ, Gawande R et al (2014) Chest CT in children: anesthesia and atelectasis. Pediatr Radiol 44:164–172
Macgowan CK, Al-Kwifi O, Varodayan F et al (2005) Optimization of 3D contrast-enhanced pulmonary magnetic resonance angiography in pediatric patients with congenital heart disease. Magn Reson Med 54:207–212
Berthezene Y, Vexler V, Clement O et al (1992) Contrast-enhanced MR imaging of the lung: assessments of ventilation and perfusion. Radiology 183:667–672
Guan Y, Xia Y, Fan L et al (2015) Quantitative assessment of pulmonary perfusion using dynamic contrast-enhanced CT in patients with chronic obstructive pulmonary disease: correlations with pulmonary function test and CT volumetric parameters. Acta Radiol 56:573–580
Calder AD, Bush A, Brody AS et al (2014) Scoring of chest CT in children with cystic fibrosis: state of the art. Pediatr Radiol 44:1496–1506
Scholz O, Denecke T, Bottcher J et al (2017) MRI of cystic fibrosis lung manifestations: sequence evaluation and clinical outcome analysis. Clin Radiol 72:754–763
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Rights and permissions
About this article

Cite this article
Amaxopoulou, C., Gnannt, R., Higashigaito, K. et al. Structural and perfusion magnetic resonance imaging of the lung in cystic fibrosis. Pediatr Radiol 48, 165–175 (2018). https://doi.org/10.1007/s00247-017-4021-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-017-4021-8