
Abstract
Pallister-Hall syndrome (PHS) is a rare condition characterised by anomalies including hypothalamic hamartoma, bifid epiglottis and postaxial polydactyly. Hearing loss has been recognised in this condition. Cochlear abnormalities have been described in mouse models of PHS, but there are no reports of similar findings in humans to date. This report describes a case of PHS with bilateral cochlear hypoplasia as seen on MRI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pallister-Hall syndrome (PHS) is a rare condition characterised by anomalies including hypothalamic hamartoma, bifid epiglottis and postaxial polydactyly. It is caused by mutations in the GLI3 zinc-finger transcription factor gene on chromosome 7p14.1. Whilst abnormal cochleae have been identified in mouse models of PHS [1], cochlear abnormalities in humans have not been described in the literature. In this report, we describe a girl with specific emphasis on hearing and the inner ear abnormality. This case has been reported previously with emphasis on genitourinary abnormalities that are unusual in PHS [2].
Case report
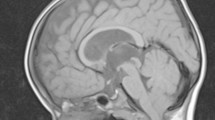
A girl born at term gestation presented with abdominal distension secondary to hydrometrocolpos and a cloacal abnormality. She was noted to have dysmorphic features including marked frontal bossing, high arched palate, anteverted nares, a flat nasal bridge and postaxial polydactyly affecting the left hand. The diagnosis of McKusick-Kaufman syndrome was considered initially. An MRI scan performed at 4 months of age investigating a probable seizure disorder revealed a large hypothalamic mass with typical characteristics of a hamartoma (Fig. 1). She was also noted to have a bifid epiglottis during induction of anaesthesia. Diagnosis of PHS was considered, based on the clinical and radiologic findings. Mutation analysis confirmed the diagnosis, revealing a nonsense mutation in the region of the GLI3 gene.

Magnetic resonance imaging (MRI) scan of the girl obtained at 6 years of age. Sagittal T1-weighted (a) and axial T2-weighted (b) images show a large hypothalamic mass (arrows) that is isointense to grey matter, demonstrating pressure effect on the midbrain and brainstem
MR imaging at 16 months suggested possible pressure effect of the hypothalamic hamartoma on the seventh and eighth cranial nerves. The imaging of the inner ears was suboptimal but revealed 1.5 turns of cochleae bilaterally and was initially thought to represent a Mondini-type abnormality.
Follow-up MR imaging of the brain at 6 years of age included the inner ears as the hearing had deteriorated considerably, according to the child’s parents. This clearly revealed the cochlea to contain 1.5 turns with bilateral absence of the apical turns (Figs. 2, 3). The remainder of the inner ear was normal and hypothalamic hamartoma remained unchanged in size with no direct pressure on the vestibulocochlear nerve.

Contiguous coronal 3-dimensional (3-D) T2 driven equilibrium (DRIVE) images reveal 1.5 turns of the cochlea on both sides. Basal turn (thin arrows) and the remaining 0.5 turn (thick arrows)

MRI images of the girl with comparative images of a normal cochlea and a cochlea with Mondini-type abnormality. Axial thick-slab T2 DRIVE image (a) shows 1.5 turns of both cochlea (arrows). This is well demonstrated on the volume-rendered comparative images of the girl’s right labyrinth (b) and a normal labyrinth (c). Selected Axial T2 DRIVE images (d) in a different patient with right sided Mondini-type cochlear abnormality demonstrates the basal turn (thin arrow) and cystic appearance of the apical and middle turns (open arrow). Normal 2.5 turns of cochlea are seen on the left side
Audiological history
There was early parental concern about hearing and a history of early hearing loss in second-degree relatives, so initial hearing assessment was carried out at 9 months of age using the click stimulus auditory brainstem response (ABR) test. This revealed moderate hearing loss with thresholds of 60 dBnHL. As a compounding factor, bilateral otitis media with effusion (OME) was identified on otoscopy and tympanometry and the hearing loss was attributed to this; hearing aids were fitted.
Subsequent audiological input was sporadic as the girl had several long inpatient episodes. More in-depth ABR testing at 3.5 years of age demonstrated absent responses to 105dBnHL, the limits of the machine, bilaterally for both mid (1,000 Hz) and high frequency (4,000 Hz) tone pips, indicating bilateral profound sensorineural hearing loss. At the age of 4 years, without regular use of hearing aids, it was noted that she could articulate words clearly, suggesting reasonable access to sound at some period in her early years and raising the possibility of additional neural component to her hearing loss. A further click stimulus ABR test at age 6 years demonstrated no clear response to 100dBnHL on either side, and cochlear microphonic was absent bilaterally, indicating that the loss was cochlear, at least in part. The developmental delay and a later diagnosis of Asperger syndrome made it difficult to obtain behavioral test results.
Discussion
Since the initial description by Hall et al. [3] in 1980, PHS has been recognised increasingly and a variety of craniofacial, cardiothoracic and genitourinary anomalies have been described in these patients in addition to the well-recognised central nervous system and limb abnormalities. Hearing loss is uncommon in PHS but has been reported. In a study by Johnson et al. [1] comprising 46 patients with PHS, 3 were reported with hearing loss. Subsequent review of this patient cohort confirmed sensorineural hearing loss in 6 out of 12 patients who had comprehensive audiological assessment. To our knowledge, radiological inner ear abnormalities in patients with PHS have not been described in the literature. Our patient had been reported previously with an emphasis on similarities with McKusick-Kaufman syndrome [2] and was thought to demonstrate Mondini-type malformation as initial scans at 16 months of age revealed 1.5 turns of the cochleae. A subsequent MRI scan at 6 years that was of better quality clearly revealed the absence of the apical turns in both cochleae. This is different from the appearances in Mondini malformation (Incomplete partition type II) where the apical and middle turns of the cochlea coalesce to form a cyst due to an interscalar defect [4] (Fig. 3). The appearances are in keeping with that of a hypoplastic cochlea as described by Giesemann et al. [5], where the cochlear size can range from a small cochlear bud to one with a normal basal turn but fewer than 2.5 turns.
Although hypoplastic cochlea have never been reported in PHS patients, Driver et al. [1] demonstrated abnormalities of the cochlea in mouse models of PHS that have appearances similar to that of our patient. In this study, Gli3Δ699/ Δ699 mice, a model for PHS was found to have cochlea that were relatively short and broad with disorganised cellular architecture when compared to the control group. Similar findings were also noted by Bok et al. [6], who postulated that Gli3 activator (GliA) and repressor (GliR) proteins are essential in the development of the ventral and dorsal structures, respectively, as a response to graded levels of exposure to Sonic Hedgehog (SHH) proteins along the dorsoventral axis during inner ear development. The distal portion of the cochlea, being the ventral-most structure of the inner ear, was deficient in the Gli3 Δ699/ Δ699 mutant mice. The abnormal Gli3 protein was thought to competitively block other Gli activators in the cochlea leading to this specific abnormality. In addition to the cochlear abnormality, our patient presented with genitourinary abnormalities, which are unusual for PHS, and also thought to be related to defects in the GLI3 and SHH pathway [2, 7].
The audiology examinations of six patients reviewed by Driver et al. [1] demonstrated mild to profound sensorineural hearing loss, which affected mainly the low frequencies. The tonotopic organisation of the cochlea with high frequency stimuli detected at the base and low frequency at the apex [8] supports this pattern of hearing loss in cochlear abnormalities closer to the apex. In our case, the audiology assessment posed a number of challenges. ABR is generated from the distal end of the cochlear nerve, via cochlear nuclei to the lateral lemniscus as a response to sound. It can provide a reliable threshold estimation for conductive and cochlear hearing loss. However, if the vestibulocochlear (eighth cranial) nerve is compromised, ABR responses quickly become absent or grossly distorted, so they are not useful for threshold estimation in hearing loss secondary to auditory neuropathy or auditory neuropathy spectrum disorder (ANSD). In our case, the first ABR at 9 months could not distinguish between conductive hearing loss secondary to OME and sensorineural hearing loss, as bone conduction ABR was not available. However, the presence of clear wave forms suggests no appreciable neural involvement at that point. The MR imaging at 16 months suggested a possible pressure effect of the hypothalamic hamartoma on the seventh and eighth cranial nerves, and it is possible that consequent auditory neuropathy affected the ABR results, making them unreliable for threshold estimation. The cochlear microphonic is generated by outer hair cells of cochlea in response to sounds. This was absent bilaterally at the age of 6 years, suggesting cochlear involvement, but it is possible that both sensory (cochlear) and neural hearing loss were present together. In summary, though there was some audiological evidence to suggest cochlear involvement, the degree of hearing loss at different frequencies caused by the cochlear abnormality could not be established in our case, because of the possible confounding effect of the pressure effect of hamartoma on the vestibulocochlear nerve, with additional intermittent conductive overlay from OME.
Conclusion
PHS is a rare syndrome with a multitude of clinical manifestations. Specific mutations in the GLI3 gene have been attributed to its etiology and have been well illustrated in animal model studies. This case illustrates the abnormalities of the cochlear apices that have been well described in mouse models of PHS but never in humans to our knowledge.
Reference
Driver EC, Pryor SP, Hill P et al (2008) Hedgehog signalling regulates sensory cell formation and auditory function in mice and humans. J Neurosci 28:7350–7358
McCann FAE, Craigie R et al (2006) Genitourinary malformations as a feature of the Pallister-Hall syndrome. Clin Dysmorphol 15:75–79
Hall JG, Pallister SK, Clarren SK et al (1980) Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus and post-axial polydactyly: a new syndrome? Part 1: clinical, causal and pathogenic considerations. Am J Med Genet 7:47–74
Sennaroglu L, Saatci I (2002) A new classification for cochleovestibular malformations. Laryngoscope 112:2230–2241
Giesemann AM, Goetz F, Neuburger J et al (2011) Appearance of hypoplastic cochlea in CT and MRI: a new classification. Neuroradiology 53:49–61
Bok J, Dolson DK, Hill P et al (2007) Opposing gradients of Gli repressor and activators mediate Shh signalling along the dorsoventral axis of the inner ear. Development 134:1713–1722
Mo R, Kim JH, Zhang J et al (2001) Anorectal malformations caused by defects in sonic hedgehog signalling. Am J Pathol 159:765–774
Davis RL (2003) Gradients of neurotrophins, ion channels, and tuning in the cochlea. Neuroscientist 9:311–316
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Avula, S., Alam, N. & Roberts, E. Cochlear abnormality in a case of Pallister-Hall syndrome. Pediatr Radiol 42, 1502–1505 (2012). https://doi.org/10.1007/s00247-012-2458-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-012-2458-3