
Abstract
Langerhans cell histiocytosis (LCH) is a disorder of unknown pathogenesis affecting one or more organs (unifocal or disseminated form) due to clonal proliferation of Langerhans cells. Liver involvement is more frequent in the disseminated form and the radiological findings of end-stage liver disease due to LCH are similar to those of sclerosing cholangitis. We present the multidetector CT findings in two children with LCH liver involvement and the unique finding of calcification of the biliary wall.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Langerhans cell histiocytosis (LCH) is a rare nonhereditary disorder of unknown aetiology, characterized by abnormal clonal proliferation of histiocytic cells phenotypically similar to Langerhans cells, and their possible accumulation in bone, skin, lymph nodes, ears, bone marrow, spleen, lungs, gastrointestinal tract, thymus, central nervous system, and liver [1]. The annual incidence of LCH is 0.5–5.4 cases per million persons per year, approximately 1 per 200,000 children per year [2]. The liver is involved in 14–19% of all affected individuals and in 54% of those with systemic LCH, particularly children younger than 2 years of age, and is regarded as a poor prognostic sign [3]. Langerhans cells infiltrate the liver directly but have a remarkable selectivity for the bile ducts leading to a sclerosing cholangiopathy (SC); the mechanism is unknown, but eventually biliary cirrhosis results [4].
We report two children, ages 6 years and 3 years, with SC secondary to LCH, referred to our institute for evaluation for liver transplantation (LT). In our practice every patient undergoes multidetector CT (MDCT) during this assessment. Images of the unenhanced, arterial and portal venous phases are acquired. Unenhanced low-dose images are acquired mainly to evaluate the unenhanced density of hepatic nodules (possible hyperdense regenerative nodules) and also to identify calcification. The arterial phase, with three-dimensional (3-D) reconstruction, is imaged to evaluate hepatic artery anatomy and to detect possible replaced or accessory hepatic arteries. The portal venous phase is imaged to evaluate the patency of the portal system, anatomical variants, such as the Abernethy malformation, and to evaluate signs suggesting the presence of portal hypertension (varices, spontaneous splenorenal shunts). As a routine, the volume of the liver is calculated as a guide for donor-to-recipient matching.
Case reports
Case 1
A 6-year-old boy had been in good health until 3 years of age. At age 4 years he presented with polyuria and diabetes insipidus was diagnosed. Clinical examination showed a firm liver with an irregular margin, palpable for approximately 5 cm below the costal margin. The spleen was also enlarged with its lower pole 12 cm below the left costal margin. Abdominal veins were visible on the anterior abdominal wall, but there was no ascites. Liver US showed a heterogeneous parenchyma with fatty infiltration and intrahepatic bile duct dilatation containing intraluminal debris. Liver biopsy confirmed infiltration by Langerhans cells. There was no evidence of bone, lymph node, skin or lung involvement.
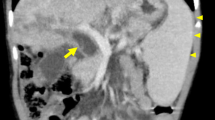
Chemotherapy was commenced with methotrexate, vinblastine and prednisone. During follow-up, a progressive reduction in serum albumin levels was associated with the development of ascites. The patient was referred to our institute and admitted with a diagnosis of SC secondary to LCH, and end-stage biliary cirrhosis. MR cholangiopancreatography (MRCP) and 64-slice MDCT were performed as part of the standard assessment prior to LT. MDCT and MRCP demonstrated cirrhotic liver morphology and severe cystic enlargement of the intrahepatic biliary ducts with intraluminal debris (Fig. 1). MDCT also showed lamellar calcification of the biliary tree wall.

Patient 1. a Unenhanced MDCT image shows curvilinear and linear calcification of the left and right main biliary ducts. Also note calcification of the right adrenal gland. b MDCT image after intravenous administration of contrast medium shows hypertrophy of the left lobe, capsular retraction of segment 4, hypertrophy of the caudate lobe, a large biliary lake with debris in the left lobe, and calcification of the wall of a right-sided bile duct. c MDCT 3-D MIP reconstruction image shows multiple bilobar calcifications of the biliary wall. Note that the right hepatic artery arises from the superior mesenteric artery. d T2-weighted MR image shows the biliary lake with debris
The patient underwent a split LT (segments 2 and 3), and the imaging findings were confirmed in the explanted liver. At the time of this report he was enjoying good health.
Case 2
A 3-year-old boy first presented at the age of 15 months with persistent fever, jaundice, lymphadenopathy, hepatomegaly (8 cm below the costal margin), splenomegaly (lower pole 12 cm below the left costal margin), and diffuse haemorrhagic lesions in the skin. Biopsies of skin and lymph nodes provided a diagnosis of LCH. Chemotherapy was commenced with methotrexate, vinblastine and prednisone, but the response was only mild. Because of increases in serum levels of gamma glutamyl transpeptidase and alkaline phosphatase on follow-up a liver biopsy was performed that showed periductal fibrosis and periportal fibrous septa with nodules, consistent with biliary cirrhosis. The boy was admitted to our institute with a diagnosis of end-stage biliary cirrhosis secondary to LCH, to be evaluated for LT.
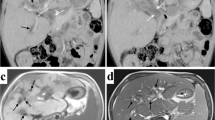
MDCT showed a liver with cirrhotic morphology, focal fatty infiltration and linear calcification of biliary walls; the biliary tree was not significantly dilated (Fig. 2). The boy underwent a split LT (segments 2 and 3) and the imaging findings were confirmed in the explanted liver.

Patient 2. a Unenhanced MDCT image shows round and linear calcification of the left and right bile ducts. b Similar findings are evident after intravenous administration of contrast medium. Note hypertrophy of the left lobe and caudate lobe
The patient died 1 year later from respiratory failure due to pulmonary reactivation of LCH.
Discussion
LCH is a disorder of histiocytic proliferation that especially affects young children. It is considered primarily to be a disorder that is reactive to infection or a consequence of abnormal immune regulation, related to cytokine and chemokine release (cytokine storm) with secondary macrophage activation syndromes and secondary haemophagocytic conditions [5]. A neoplastic origin is also suggested by the clonality of the LCH cells; however, the pathogenesis is not yet clear [6].
LCH is staged according to the number of systems involved and the presence of organ dysfunction. Multisystem LCH is characterized by granulomatous infiltration of lung, bone, skin, lymph nodes, liver, spleen, brain, kidneys, and the endocrine system, and can be categorized as LCH “without organ dysfunction” and LCH “with organ dysfunction”. Multiorgan involvement and age younger than 2 years are predictors of decreased survival, while patients with solitary bone lesions have a good prognosis.
Patients with hepatic involvement and dysfunction usually have the disseminated form of the disease with a poor prognosis. In patients with liver involvement, the 3-year survival rate is 51.8%, compared to 96.7% in patients without liver involvement [4]. Liver involvement manifests with clinical and biochemical signs: hepatomegaly, splenomegaly secondary to portal hypertension, hypoalbuminaemia, coagulopathy, obstructive jaundice, and elevated gamma glutamyl transpeptidase or alkaline phosphatase secondary to direct bile duct infiltration [4, 6, 7].
The imaging findings of liver involvement depend on which of the four progressive histological phases is present: proliferative, granulomatous, xanthomatous, or the final fibrous phase [5, 7]. The proliferative and granulomatous phases are characterized by periportal histiocyte infiltration, inflammation and oedema, which can appear as periportal oedema that is hypoechoic on US images, hypodense on CT images, hypointense on T1-W MR images and hyperintense on T2-W MR images. Portal triaditis has been reported; it shows periportal areas of low attenuation on CT images following contrast medium injection. In the xanthomatous phase, periportal fatty lesions can be seen. These appear hyperechoic on US images, hypodense on CT images, high signal on T1-W MR images, and low signal on T2-W MR images. The final stage is characterized by predominant periductal fibrosis and micronodular biliary cirrhosis. The fibrotic disease process can lead to SC (in 10–15% of patients with multisystem LCH with a median onset 2 years after diagnosis) and biliary cirrhosis [3, 7–11]. The usual findings of SC are multiple and irregular biliary strictures and dilatations. Beaded biliary duct enlargement or biliary lakes with stones have been described in adults, but not in children.
Spotty calcification in the thymus and in the liver has been described in LCH, but hepatic calcification appears to be very unusual [11–13]. Both our patients showed advanced involvement of the liver with biliary cirrhosis secondary to SC. Patient 1 showed dilated and beaded bile ducts with stones, while patient 2 did not show significant biliary dilatation. Accumulation of tiny calcified concretions in areas of cellular necrosis have been described as a possible explanation for punctuate thymic calcifications in patients with LCH [12] and a similar mechanism could be the basis of the biliary wall calcifications seen in our two patients, although a contribution from the chemotherapy cannot be excluded. In conclusion lamellar biliary wall calcification can be present in children with LCH. It is not possible to conclude whether this finding is related to staging or prognosis.
References
Alston RD, Tatevossian RG, McNally RJ et al (2007) Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer 48:555–560
Broadbent V, Egeler RM, Nesbit ME Jr (1994) Langerhans’ cell histiocytosis – clinical and epidemiological aspects. Br J Cancer 70 [Suppl XXIII]:S11
Braier J, Ciocca M, Latella A et al (2002) Cholestasis, sclerosing cholangitis and liver transplantation in Langerhans cell histiocytosis. Med Pediatr Oncol 38:178–182
Wong A, Ortiz-Neira CL, Reslan WA et al (2006) Liver involvement in Langerhans cell histiocytosis. Pediatr Radiol 36:1105–1107
Jaffe R (2004) Liver involvement in the histiocytic disorders of childhood. Pediatr Dev Pathol 7:214–225
Nezelof C, Basset F (2004) An hypothesis Langerhans cell histiocytosis: the failure of the immune system to switch from an innate to an adaptive mode. Pediatr Blood Cancer 42:398–400
Kilborn TN, Teh J, Goodman TR (2003) Paediatric manifestations of Langerhans cell histiocytosis: a review of the clinical and radiological findings. Clin Radiol 58:269–278
Pagnoux C, Hayem G, Roux F et al (2003) Sclerosing cholangitis as a complication of Langerhans’ cell histiocytosis. Rev Med Interne 24:324–327
Kim M, Lyu C, Jin Y et al (1999) Langerhans’ cell histiocytosis as a cause of periportal abnormal signal intensity on MRI. Abdom Imaging 24:373–377
Chaudhary A, Debnath J, Thulkar S et al (2006) Imaging findings in hepatic Langerhans’ cell histiocytosis. Indian J Pediatr 73:1036–1038
Buza N, Lagarde DC, Dash S et al (2004) Langerhans cell histiocytosis: report of a single organ involvement in a child. J Cell Mol Med 8:397–401
Heller GD, Haller JO, Berdon WE et al (1999) Punctate thymic calcification in infants with untreated Langerhans’ cell histiocytosis: report of four new cases. Pediatr Radiol 29:813–815
Arakawa A, Tetsuya M, Yamashita Y et al (1994) Periportal fibrosis in Langerhans’ cell histiocytosis mimicking multiple liver tumors: US, CT and MR findings. J Comput Assist Tomogr 18:157–159
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Caruso, S., Miraglia, R., Maruzzelli, L. et al. Biliary wall calcification in Langerhans cell histiocytosis: report of two cases. Pediatr Radiol 38, 791–794 (2008). https://doi.org/10.1007/s00247-008-0809-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-008-0809-x