
Abstract
Langerhans cell histiocytosis (LCH) is a rare, proliferative disorder that occurs primarily in children. Periportal lesions were detected on ultrasound at different stages of evolution. In this article, we aim to discuss the sonographic findings in liver involvement of LCH.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Langerhans cell histiocytosis, formerly known as histiocytosis X, may present as a multisystemic disease [1].
In this context, a rare case of hepatic involvement in Langerhans cell histiocytosis detected with ultrasonography is presented in this report. Various radiologic modalities, including ultrasonography, contrast-enhanced tomography (CT), and magnetic resonance imaging (MRI), can detect hepatic involvement in Langerhans cell histiocytosis. However, among these techniques, CT requires radiation exposure, and MRI often requires sedation. Therefore, the initial imaging modality to be used in infants and neonates should be ultrasonography, whereas MRI can be used complementarily if needed for clinical management. In parallel, grayscale ultrasound and color Doppler ultrasound examinations were performed using an Aplio i800 ultrasound scanner (Canon Medical Systems USA, Tustin, CA) equipped with convex 8 MHz (i8CX1, Aplio i800TM, Canon Medical Systems) and linear array 18 MHz (i18LX5, Aplio i800TM, Canon Medical Systems) transducers.
Case report
A 3-year-old girl presented to the pediatric department with a three-month history of intermittent abdominal pain and nausea. Her clinical examination revealed scleral icterus and marked hepatomegaly, whereas her laboratory tests carried out at admission revealed elevated liver function results. The patient had a history of Langerhans cell histiocytosis for two years.
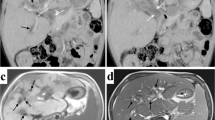
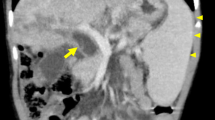
She was referred to the radiology department for further evaluation. Grayscale ultrasound indicated extensive, linear periportal hyperechogenic lesions and hepatomegaly (Fig. 1A). No dilatation was observed in the intrahepatic bile ducts. The Doppler study did not reveal any vascularity in the periportal lesions (Fig. 1B). Based on imaging findings and her clinical history, the patient was pre-diagnosed with hepatic involvement in Langerhans cell histiocytosis. The histopathology of the tru-cut biopsy specimen supported the preliminary diagnosis. In line with this diagnosis, the patient was started on immunosuppressives and a combined chemotherapy regimen. The results of the MRI performed four months after the start of the treatment did not reveal any regression of periportal lesions and focal dilatation of the peripheral bile duct (Fig. 2). Periportal lesions appeared as hyperintensity on axial T2-weighted image, hypointensity on T1 weighted image, and no contrast enhancement was observed in contrast-enhanced fat-suppressed T1 images MRI. Bone marrow and liver transplants have been planned based on multidisciplinary consensus.

A 3-year-old girl with Langerhans cell histiocytosis. Histopathologically, periportal Langerhans cell infiltration with features of granulomatous and xanthomatous phases is observed in the liver. Grayscale ultrasound revealed A extensive hyperechogenic areas along the portal triad (red arrow) and hepatomegaly with the convex probe on the transverse section of the liver. B No vascularity of periportal lesions in the color Doppler study

MRI images of the liver in a hepatic LCH case. A A follow-up axial T2-weighted image revealed periportal high signal intensity (red arrow) and slight focal dilatation of the peripheral bile duct (black arrow). B and C No contrast enhancement was observed on pre- and post contrast T1 images
Discussion
Langerhans cell histiocytosis (LCH) is a rare, proliferative disorder that occurs primarily in children. LCH is characterized by granulomatous infiltration. The unique pathological feature of LCH is an inappropriate proliferation of abnormal Langerhans cells, which may infiltrate almost any tissue. The underlying etiology of LCH is unknown [1].
Common sites of involvement are the bone marrow, central nervous system, lung, skin, spleen, and lymph nodes. Extraosseous involvement is less commonly seen than osseous involvement. Disseminated LCH affects multiple organs and may lead to a fatal outcome. More than 90% of LCH patients with multisystem involvement are children. The clinical presentation depends on the extent of organ involvement. Hence, LCH can present in various forms, ranging from localized bone lesions to disseminated lesions [2].
Hepatic involvement occurs mainly in disseminated LCH. Clinical characteristics of LCH with hepatic involvement are hepatomegaly, jaundice, and liver function dysfunction, in addition to periportal abnormalities, which are characteristic of hepatic infiltration in LCH.
Radiological findings reflect the underlying histopathological process, which consists of four phases: the proliferative, granulomatous, xanthomatous, and final fibrous phases. Periportal infiltration by histiocytes is observed as linear hypoechogenicity in ultrasonography, hypodense in CT, and moderate to high signal intensity in T2-weighted image MRI in the proliferative and granulomatous phases. Periportal contrast enhancement may also be present. The xanthomatous phase is characterized by lipid-laden nodules, which are observed as hyperechoic in ultrasonography, hypodense in CT, and hyperintense in unenhanced T1-weighted image MRI without fat suppression. The fibrous phase is characterized by concentric periductal fibrosis, which leads to biliary irregularities with segmental stenosis and dilatation. Periportal lesions appear with low signal intensity on T2-weighted MRI scans in the fibrous phase. The final clinical picture is liver failure. Periportal changes and imaging features are not pathognomonic and thus should be supported by clinical history and histopathology [3, 4].
The clinical prognosis varies depending on the disease burden and the degree of organ dysfunction at the time of the initial diagnosis. Hepatic involvement is associated with a poor prognosis. The treatment approaches generally include the use of corticosteroids and multiagent chemotherapy, though a standardized treatment approach has not been developed yet. Response to the treatment is unpredictable, and progression can occur after the treatment. In the final stage, hepatic transplantation is the only curative solution [1].
In sum, this case report demonstrated the importance of using ultrasonography for following up on patients with Langerhans cell histiocytosis. In addition, MRI can be used for further evaluation of the biliary ducts. Radiologists must be familiar with the sonographic appearance of the hepatic involvement of Langerhans cell histiocytosis for early diagnosis. Early and effective diagnosis is crucial for preventing periductal fibrosis and liver failure.
References
Schmidt S, Eich G, Geoffray A, Hanquinet S, Waibel P, Rainer WR et al (2008) Extraosseous Langerhans cell histiocytosis in children. Radiographics 28(3):707–726
Kilborn TN, Teh J, Goodman TR (2003) Paediatric manifestations of Langerhans cell histiocytosis: a review of the clinical and radiological findings. Clin Radiol 58(4):269–278
Shi Y, Qiao Z, Xia C, Gong Y, Yang H, Li G et al (2014) Hepatic involvement of Langerhans cell histiocytosis in children imaging findings of computed tomography, magnetic resonance imaging and magnetic resonance cholangiopancreatography. Pediatr Radiol. 44(6):713–718
Chan YL, Li CK, Lee CY (1997) Sonographic appearance of hepatic Langerhans cell histiocytosis. Clin Radiol 52:761–763
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. The study protocol was approved by the local ethics committee.
Informed consent
Approval from the Institutional Review Board was obtained, and in keeping with the policies for a retrospective review, informed consent was not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tezel, Z.E., Keven, A. & Mersinlioğlu, İ. Hepatobiliary involvement in Langerhans cell histiocytosis. J Ultrasound 27, 169–171 (2024). https://doi.org/10.1007/s40477-023-00811-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40477-023-00811-6