
Abstract
Barth syndrome is an X-linked disorder characterized by dilated cardiomyopathy, cyclic neutropenia, skeletal myopathy, abnormal mitochondria, and growth deficiency. The primary defect is a mutation in the TAZ gene on the X chromosome at Xq28, resulting in abnormal phospholipid biosynthesis and cardiolipin deficiency. To date, there has been no systematic evaluation of the cardiac phenotype. We report five cases of cardiac arrest and/or placement of an internal cardiac defibrillator with documented ventricular arrhythmia. We suggest that ventricular arrhythmia is part of the primary phenotype of the disorder and that patients should be screened accordingly.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Barth syndrome is an X-linked metabolic disorder first fully described in 1983 [1]. The syndrome is characterized by dilated cardiomyopathy (DCM), skeletal myopathy, neutropenia, short stature, and 3-methylglutaconic aciduria. Death in childhood is usually from sepsis or heart failure, although sudden death has been noted [2]. The disease course can be variable, even within families. While rare, Barth syndrome may be underdiagnosed and accurate incidence rates are unavailable. There are currently 69 living and 62 deceased patients in the Barth Syndrome Foundation registry, and 80 cases have been genotyped by the primary lab.
Most boys present in infancy with DCM, although presentation with delayed motor milestones or recurrent infection has been reported. Clinical cardiac features reported in the literature have included left ventricular (LV) dilation and hypertrophy with varying degrees of congestive heart failure. Left ventricular noncompaction has been reported, and autopsy reports have indicated endocardial fibroelastosis in some cases [1, 16]. Although no description of the natural history has been published, some clinicians have noted improvement in the cardiac function over time in patients who survive past infancy.
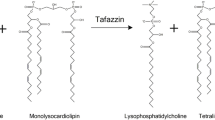
Early reports suggested a mitochondrial defect with respiratory chain abnormalities in skeletal muscle mitochondria and abnormal architecture of mitochondria in cardiac muscle [1, 15]. The gene for Barth syndrome was mapped to Xq28 [4], and the TAZ gene was subsequently cloned [3] and characterized [5, 10]. Mutations at this locus cause not only classic Barth syndrome but also other forms of cardiomyopathy (DCM and LV noncompaction) [5]. It is known that, via alternative splicing and initiation sites, this gene encodes a family of proteins, termed “tafazzins,” that have a shared homology with the acyltransferases involved in phospholipid biosynthesis [17]. Cardiolipin (CL), a mitochondrial membrane lipid, is deficient in Barth syndrome patients [19]. More specifically, incorporation of linoleic acid into CL is significantly reduced [19], resulting in near-absence of tetralinoleoyl–cardiolipin [18], a CL subclass. This specific mitochondrial phospholipid species is enriched in normal skeletal and cardiac muscle [18]. Many unique mutations in the TAZ gene have been described, and no genotype–phenotype correlation has been demonstrated [10].
We report five cases of ventricular arrhythmia leading to cardiac arrest or the placement of an internal cardiac defibrillator (ICD), with the majority of cases in this series occurring in adolescents or young adults. All patients were known to have Barth syndrome prior to documentation of arrhythmia. No cases occurred during intercurrent illnesses, and all QTc intervals were normal.
Cases
All Five Cases are Summarized in Table 1.
Case 1
A 13-year-old boy with mild DCM on digoxin [most recent echo demonstrated stable shortening fraction (SF) of 31% 8 months prior to cardiac arrest] collapsed suddenly on a playground. He was found by paramedics to have ventricular fibrillation and was successfully defibrillated. Postarrest, the SF was 18%, serum electrolytes were normal, and the white blood cell count (WBC) was 1500/μl. Left ventricular end-diastolic dimension (LVEDd) was 4.38 cm (Z-score = 2.4), with no significant interval change from the previous study. An electrocardiogram (ECG) demonstrated normal sinus rhythm (NSR) with normal conduction intervals (QTc = 413 msec). Brain imaging and electroencaphalogram (EEG) were consistent with severe hypoxic/ischemic injury. Support was withdrawn and the patient died.
Previous cardiac history included stable LV dimensions and systolic function and normal conduction intervals by ECG. A 24-hour Holter monitor performed 18 months prior to death revealed a single premature ventricular contraction (PVC) without other arrhythmia, and ST segment depression was present at elevated heart rates. The patient history was remarkable for episodes of pallor, nausea, and chest pain not associated with exercise or palpitations that were thought to be consistent with vasovagal symptoms. Transtelephonic recordings during these episodes revealed no arrhythmia. Family history was remarkable for sudden death while playing in a 3-½-year-old brother with DCM, skeletal myopathy, and neutropenia consistent with Barth syndrome.
Case 2
An 11-year-old boy with mild DCM (most recent SF was 29% 11 months prior to cardiac arrest) collapsed suddenly at home while playing. CPR was initiated, Torsades de pointes was recorded, and he was successfully cardioverted. On admission, serum electrolytes were normal and WBC was 2100/μl. On echocardiography, SF was 24% and LVEDd was 4.7 cm (Z-score = 2.8). An ECG demonstrated NSR, inverted T-waves in the lateral precordial leads, and a QTc of 444 msec. Invasive electrophysiology (EPS) without pharmacologic stimulation performed 7 days after cardiac arrest revealed sustained polymorphic ventricular tachycardia (VT) with triple extrastimulation in the RV apex (cycle length = 400/240/180/180 msec). A single-lead ICD was placed. Medications at the time of admission included digoxin, enalapril, vitamin E, coenzyme Q, vitamin C, carnitine, cisapride, and ranitidine. Cisapride had been started >10 months earlier, and ECG demonstrated normal conduction intervals after its initiation. The patient history included ECG with normal conduction intervals, a 24-hour Hotter several years prior without evidence of arrhythmia, echocardiograms with stable ventricular dimensions and systolic function, and clinical symptoms consistent with vasovagal physiology (dizziness, nausea, chest pain, fatigue). Family history was negative.
Three years after ICD implantation, a Holter monitor revealed an eight-beat run of VT. Four years after implantation, the patient had a discharge from the ICD. Investigation revealed a narrow QRS tachycardia with a cycle length of 240 msec that was successfully converted to NSR. An EPS revealed atypical atrioventricular (AV) node reentry tachycardia. The patient underwent successful slow pathway modification. An aggressive ventricular stimulation protocol was not performed to determine the inducibility of ventricular tachydysrhythmias. Subsequently, he had an episode of ventricular fibrillation during active play and was successfully defibrillated by a single shock from the ICD.
Case 3
A 16-year-old boy with stable, mildly reduced ventricular systolic function (SF = 25%–30%) developed palpitations but no syncope. Multiple ECGs demonstrated NSR with stable right bundle branch (RBBB) pattern. Medications included digoxin and lisinopril. Holter monitor revealed frequent uniform PVCs (∼ 50/hour) with couplets. A microvolt T-wave alternans (TWA) test was positive with sustained alternans at a heart rate of 104. The patient was placed on atenolol. Follow-up Holter revealed fewer couplets but no overall change in the total PVCs, and follow-up TWA was equivocal. Exercise treadmill test with a Bruce protocol was performed; total exercise time was 4.5 minutes, with a maximum heart rate of 87% predicted. Uniform PVCs improved with exercise and returned in early recovery including a three-beat run of VT. Holter monitor performed 3 months after initiation of atenolol revealed frequent PVCs, couplets, and triplets.
Four months after initiation of atenolol therapy, the patient presented with chest pain and increased palpitations. Cardiac enzymes were normal and repeat TWA on higher-dose atenolol was positive with sustained alternans at a heart rate of 97. An EPS without pharmacologic stimulation revealed normal sinus and AV node function, multifocal ventricular ectopy, and induction of reproducible VF with triple extrastimuli (cycle length = 600/290/220/220 msec) in the RV outflow tract, requiring defibrillation. An ICD was placed.
Other history included episodes of fatigue, nausea, and pallor consistent with vasovagal physiology that were significantly improved with fludrocortisone. Family history was remarkable for the sudden death of a 4-year-old brother with DCM and findings consistent with Barth syndrome.
Case 4
An 18-year-old boy with DCM, poor but stable LV systolic function [ejection fraction (EF) = 20%] and LV morphology consistent with LV noncompaction had palpitations and two episodes of syncope. Electrocardiograms demonstrated NSR with normal conduction intervals (QTc = 410–420) and left ventricular hypertrophy (LVH) with strain. Medical management included digoxin, lisinopril, coumadin, and granulocyte colony–stimulating factor (GCSF). Holter monitor revealed nonsustained ventricular tachycardia (NSVT), and aminodarone was initiated.
Subsequently, the patient suffered syncope and had a cerebrovascular accident (CVA), resulting in right hemiparesis that was treated with thrombolytic therapy. Echocardiography revealed LV thrombus, which was treated with anticoagulation and resolved on follow-up studies. Due to the history of a recent CVA, an ICD was placed without an EPS. Subsequent interrogation has revealed no arrhythmia.
Additional history included episodes consistent with vasovagal physiology and a positive tilt-table test with syncope. Family history was remarkable for a brother with Barth syndrome, another brother who died at 7 months of age from respiratory symptoms, and a maternal nephew with Barth syndrome who died during a hospitalization with croup.
Case 5
A 17-year-old boy with stable ventricular systolic function (SF = 32%–34%) without history of syncope or significant palpitations had normal 24-hour Holter and signal-averaged EGG (SA-ECG) evaluations. He had an EPS performed to evaluate the risk of life-threatening arrhythmia, given the documented arrhythmia in other Barth syndrome patients. This EPS without pharmacologic stimulation demonstrated normal sinus and AV node function with easily reproducible sustained polymorphic VT with double and triple stimuli in the RV apex. An ICD was placed, and no discharges occurred in the first 11 months after implantation. Medical management included enalapril, carvedilol, digoxin, carnitine, coenzyme Q, and GCSF. Prior ECG revealed diffusely flat ST segments with normal conduction intervals (QTc = 405 msec).
Additional history included severe chronic fatigue and vasovagal symptoms (episodic dizziness, abdominal pain, headache with normal transtelephonic monitoring studies during the symptoms). Family history was negative.
Discussion
Barth syndrome is an inherited cardiomyopathy arising from a single gene defect. The cases reported here suggest that ventricular arrhythmia may be an important part of the pathophysiology, morbidity, and mortality. Of our five patients with documented ventricular arrhythmia, four had mild LV dilation with low normal or mildly depressed LV systolic function, and only one had very poor but stable systolic function prior to cardiac arrest or ICD placement. This suggests that the risk of arrhythmia may be independent of the degree of LV dilation or dysfunction. Each of these patients was treated differently with regard to screening for rhythm disturbance. All of these five patients had multiple ECGs with normal conduction intervals. Two had a positive arrhythmia screening study (Holter and/or TWA), one had repolarization abnormalities at elevated heart rates on Holter but no documented arrhythmia, one had a normal Holter, and one had no recent Holter or other arrhythmia screening tests. Three of the five patients had EPS, all of which demonstrated inducible ventricular arrhythmia, and two patients had syncope prior to ICD placement or cardiac arrest. In addition, one patient had both ventricular and supraventricular tachycardia. It is interesting to note that all patients in this group had significant and recurrent vasovagal symptoms, with nausea, pallor, and dizziness suggestive of autonomic dysfunction. Thus, in some cases, it may be difficult to distinguish symptomatic arrhythmia from vasovagal physiology.
We have had two additional cases of ICD placement in patients with Barth syndrome for sudden cardiac death prophylaxis. One of these patients was the 21-year-old brother of case 4 and was found to have mild but stable cardiomyopathy (SF = 27%) and concentric LVH by echocardiography. Symptoms included palpitations, presyncope, and one episode of syncope. Given the clinical history of cardiomyopathy and palpitations with a family history of arrhythmia and death, an ICD was placed as a primary measure to prevent sudden death. No EPS was performed. The second additional case was in a 21-year-old patient with DCM and was managed with digoxin, lasix, captopril, and metoprolol. There was no history of palpitations or syncope. The LV dimensions and systolic function have been stable (SF = 30%). Although no EPS was performed, an ICD was placed for primary prevention of sudden death, given the chronic DCM and known serious arrhythmia in other patients with Barth syndrome.
It is not unexpected that patients with Barth syndrome have an increased risk of arrhythmia. In addition to the underlying substrate of DCM, there is potential for cardiac rhythm disturbance related to abnormalities of mitochondrial function. There are two possible mechanisms for the observed rhythm abnormalities. One involves adenosine triphosphate (ATP)–sensitive K+ (KATP) channels in the inner mitochondrial membrane of cardiomyocytes, which affect intramitochondrial depolarization and have important cardioprotective effects during ischemia and ischemia-related arrhythmia [12]. Altered fatty acid distribution of the mitochondrial membrane phospholipids may disturb lipid–protein interactions and alter mitochondrial ion channels, thus predisposing to arrhythmia. Another potential mechanism involves the role that CL plays in apoptosis. Cardiolipin is a phospholipid located solely on the inner mitochondrial membrane and is nearly absent in patients with Barth syndrome [18, 19]. Cardiolipin tightly binds cytochrome C, and release of this cytochrome from the mitochondrial membrane into the cell is an initial step in programmed cell death [13]. Cardiolipin is also required for adenine nucleotide translocase (ANT), which is important in mitochondrial permeability [13]. Cardiolipin deficiency is related to increased mitochondrial membrane permeability and release of cytochrome C and other cell death mediators into the cell, thus inducing apoptosis. Programmed cell death has been associated with cardiomyopathy and cardiac rhythm disturbance [13, 14].
Despite significant progress in the diagnosis and management of arrhythmia in patients with cardiomyopathy and heart failure, the prediction of patients at risk for sudden cardiac death has proven extremely difficult. There is very little information regarding risk stratification in children, and the current data in the literature are from the adult population. The Marburg Cardiomyopathy Study [8] evaluated measures from echocardiography, signal-averaged ECG, Holter, baroreflex sensitivity, and TWA testing in idiopathic DCM in patients aged 16–70 years over 51 ± 21 months. This study found that LV EF <30%, lack of beta-blocker therapy, or the combination of LV EF <30% with NSVT on Holter were the only significant predictors of major arrhythmic events. However, other authors have suggested that individual arrhythmia screening studies may be useful in predicting arrhythmia events, For example, the presence of NSVT on 24-hour Holter monitor has been shown to be a risk factor for sudden death in adult patients with severe congestive heart failure [6]. Both signal-averaged ECG [7] and TWA [9] have also been shown to have positive predictive power in DCM. Autonomic dysfunction may also play a role in arrhythmia, and heart rate variability has been shown to be a predictor of sudden death in adult patients with CHF [11]. This is especially intriguing given the symptoms of autonomic dysfunction that appear to be present in these patients with Barth syndrome.
Although Barth syndrome is a rare form of DCM, it may be underdiagnosed in boys with cardiomyopathy. These cases suggest that life-threatening arrhythmia independent of the degree of cardiac dilation or dysfunction may be an important clinical manifestation of this disorder. Of note, two of the five cases of proven arrhythmia had a family history of sudden cardiac death in a brother with probable Barth syndrome, and one had a family history of death that may have been due to sudden cardiac arrest. There was no demonstrable genotype–phenotype correlation between the type of mutation and the presence of arrhythmia.
Although the literature indicates that screening for cardiac rhythm abnormality in idiopathic DCM is not particularly sensitive or specific, we think that patients with Barth syndrome may represent a special subset of patients with DCM and an inherent risk of ventricular arrhythmia. We believe that they should have an intensified approach to screening for life-threatening arrhythmia, especially if there is a positive family history of cardiac arrest. Standardized cardiac assessment, including (at a minimum) yearly echocardiography, EGG, and Holter monitoring, should be considered for patients with Barth syndrome. In addition, consideration should be given to enhanced arrhythmia screening, including exercise testing, SA-ECG, and TWA. It may be prudent to have a low threshold for performing an EPS to investigate potentially serious arrhythmia, especially in the presence of symptoms, abnormal arrhythmia screening tests, or a family history of sudden death. There are no data on the use of antiarrhythmic medication in this patient population. Further investigation into the mechanism of the disease and arrhythmia is warranted.
References
PG Barth HR Scholte JA Berden et al. (1983) ArticleTitleAn X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes J Neurol Sci 62 327–355 Occurrence Handle6142097 Occurrence Handle10.1016/0022-510X(83)90209-5 Occurrence Handle1:STN:280:BiuC3sjntFI%3D
PG Barth RJ Wanders P Vreken et al. (1999) ArticleTitleX-linked cardioskeletal myopathy and neutropenia (Barth syndrome) (MIM 302060) J Inherit Metab Dis 22 555–567 Occurrence Handle10407787 Occurrence Handle10.1023/A:1005568609936 Occurrence Handle1:CAS:528:DyaK1MXkslOmtLk%3D
S Bione P D’Adamo E Maestrini et al. (1996) ArticleTitleA novel X-linked gene, G4.5. is responsible for Barth syndrome Nat Genet 12 385–389 Occurrence Handle8630491 Occurrence Handle10.1038/ng0496-385 Occurrence Handle1:CAS:528:DyaK28XjtFaju7Y%3D
PA Bolhuis GW Hensels TJ Hulsebos F Baas PG Barth (1991) ArticleTitleMapping of the locus for X-linked cardioskeletal myopathy with neutropenia and abnormal mitochondria (Barth syndrome) to Xq28 Am J Hum Genet 48 481–485 Occurrence Handle1998334 Occurrence Handle1:STN:280:By6C2M%2FnsFM%3D
P D’Adamo L Fassone A Gedeon et al. (1997) ArticleTitleThe X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies Am J Hum Genet 61 862–867 Occurrence Handle9382096 Occurrence Handle1:CAS:528:DyaK2sXmslCns7k%3D
HC Doval DR Nul HO Grancelli et al. (1996) ArticleTitleNonsustained ventricular tachycardia in severe heart failure: independent marker of increased mortality due to sudden death. GESICA–GEMA investigators Circulation 94 3198–3203 Occurrence Handle8989129 Occurrence Handle1:STN:280:ByiC2czhtFw%3D
L Fauchier D Babuty P Cosnay et al. (2000) ArticleTitleLong-term prognostic value of time domain analysis of signal-averaged electrocardiography in idiopathic dilated cardiomyopathy Am J Cardiol 85 618–623 Occurrence Handle11078277 Occurrence Handle10.1016/S0002-9149(99)00821-8 Occurrence Handle1:STN:280:DC%2BD3M%2FkslCmtA%3D%3D
W Grimm M Christ J Bach HH Muller B Maisch (2003) ArticleTitleNoninvasive arrhythmia risk stratification in idiopathic dilated cardiomyopathy: results of the Marburg Cardiomyopathy Study Circulation 108 2883–2891 Occurrence Handle14623812 Occurrence Handle10.1161/01.CIR.0000100721.52503.85
SH Hohnloser T Klingenheben D Bloomfield O Dabbous RJ Cohen (2003) ArticleTitleUsefulness of microvolt T-wave alternans for prediction of ventricular tachyarrhythmic events in patients with dilated cardiomyopathy: results from a prospective observational study J Am Coll Cardiol 41 2220–2224 Occurrence Handle12821251 Occurrence Handle10.1016/S0735-1097(03)00467-4
J Johnston RI Kelley A Feigenbaum et al. (1997) ArticleTitleMutation characterization and genotype–phenotype correlation in Barth syndrome Am J Hum Genet 61 1053–1058 Occurrence Handle9345098 Occurrence Handle10.1086/301604 Occurrence Handle1:CAS:528:DyaK2sXns1yjuro%3D
MT La Rovere GD Pinna R Maestri et al. (2003) ArticleTitleShort-term heart rate variability strongly predicts sudden cardiac death in chronic heart failure patients Circulation 107 565–570 Occurrence Handle12566367 Occurrence Handle10.1161/01.CIR.0000047275.25795.17
Y Liu T Sato B O’Rourke E Marban (1998) ArticleTitle(1998) Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation 97 2463–2469 Occurrence Handle9641699 Occurrence Handle1:CAS:528:DyaK1cXkt1Gis7g%3D
JB McMillin W Dowhan (2002) ArticleTitleCardiolipin and apoptosis Bioohim Biophys Acta 1585 97–107 Occurrence Handle1:CAS:528:DC%2BD3sXivVOqsA%3D%3D
P Nerheim SC Krishnan B Olshansky K Shivkumar (2001) ArticleTitleApoptosis in the genesis of cardiac rhythm disorders Cardiol Clin 19 155–163 Occurrence Handle11787809 Occurrence Handle10.1016/S0733-8651(05)70201-0 Occurrence Handle1:STN:280:DC%2BD38%2FmtFaksQ%3D%3D
HB Neustein PR Lurie B Dahms M Takahashi (1979) ArticleTitleAn X-linked recessive cardiomyopathy with abnormal mitochondria Pediatrics 64 24–29 Occurrence Handle572031 Occurrence Handle1:STN:280:CSaB3c%2Fnslw%3D
HB Neustein PR Lurie M Fugita (1979) ArticleTitleEndocardial fibroelastosis found on transvascular endomyocardial biopsy in children Arch Pathol Lab Med 103 214–219 Occurrence Handle582252 Occurrence Handle1:STN:280:CSaB3cfks1A%3D
AF Neuwald (1997) ArticleTitleBarth syndrome may be due to an acyltransferase deficiency Curr Biol 7 R465–R466 Occurrence Handle9259571 Occurrence Handle10.1016/S0960-9822(06)00237-5 Occurrence Handle1:CAS:528:DyaK2sXlt12ktrc%3D
M Schlame JA Towbin PM Heerdt et al. (2002) ArticleTitleDeficiency of tetralinoleoyl–cardiolipin in Barth syndrome Ann Neurol 51 634–637 Occurrence Handle12112112 Occurrence Handle10.1002/ana.10176 Occurrence Handle1:CAS:528:DC%2BD38XjvFGhs7s%3D
P Vreken F Valianpour LG Nijtmans et al. (2000) ArticleTitleDefective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome Biochem Biophys Res Commun 279 378–382 Occurrence Handle11118295 Occurrence Handle10.1006/bbrc.2000.3952 Occurrence Handle1:CAS:528:DC%2BD3cXovVCnt78%3D
Acknowledgment
We thank Iris Gonzalez, Ph.D., for the mutation analysis. We are also grateful to the families of these patients for their willingness to contribute. C.T.S. is a recipient of the Howard Hughes Medical Institutes Award for faculty development.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Spencer, C., Byrne, B., Gewitz, M. et al. Ventricular Arrhythmia in the X-linked Cardiomyopathy Barth Syndrome. Pediatr Cardiol 26, 632–637 (2005). https://doi.org/10.1007/s00246-005-0873-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-005-0873-z