
Abstract
Ambient air polycyclic aromatic hydrocarbon (PAH) samples were collected at traffic, residential, coastal, and semiurban sites in Bursa, Turkey, between June 2008 and June 2009. For the traffic, residential, coastal, and semiurban sites, the average gas phase total PAH (∑12PAH) concentrations were 113 ± 131, 142 ± 204, 53 ± 73, and 19 ± 34 ng/m3, respectively, whereas the average particle phase total PAH concentrations were 28 ± 36, 56 ± 85, 24 ± 40, and 11 ± 23 ng/m3, respectively. Phenanthrene and fluoranthene had the highest concentrations of all of the sampling sites in the gas phase. The PAH concentrations in the heating period were 5–7 times greater than the nonheating period concentrations. Principal component analysis (PCA) was used to investigate the relationship between the levels of PAHs determined in ambient samples and their possible sources. The PCA model shows that coal combustion and vehicle emissions affected PAH emissions. Moreover, the molecular diagnostic ratios indicated that coal-burning and traffic emissions were the dominant PAH sources. The multiple linear regression analysis indicated that the meteorological parameters also affected the ambient PAH concentrations. The sampling site characteristics, meteorological conditions, dispersion, and local sources all affected the concentration levels.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Polycyclic aromatic hydrocarbons (PAHs) are a class of complex organic chemicals that include carbon and hydrogen with a fused ring structure containing at least two benzene rings (World Health Organization 1987; Agency For Toxic Substances And Disease Registry 1994; Ravindra et al. 2008). PAHs are widely distributed in the atmosphere and were one of the first atmospheric pollutants to be identified as a suspected carcinogen (International Agency for Research on Cancer 1983). As the molecular weight (MW) increases, the carcinogenicity of PAHs also increases and the acute toxicity decreases (Ravindra et al. 2001).
In general, there are five major PAH emission sources including domestic, mobile, industrial, agricultural, and natural sources (Wing and Bada 1992; Beegle et al. 2001; Halasinski et al. 2005). PAHs emitted to the atmosphere are mostly formed during the incomplete combustion and pyrolysis of fossil fuels or wood and from the release of petroleum products (Ding et al. 2007; Rajput and Lakhani 2010). Other sources of PAHs include petroleum spills, oil seepage, and diagenesis of organic matter in anoxic sediments (Rubailo and Oberenko 2008; Inomata et al. 2012). PAHs are also found in coal tar, crude oil, creosote, and roofing tar, and a few are used in medicine or to make dyes, plastics, and pesticides (Simcik et al. 1998; Mastral and Callen 2000; Lim et al. 2005; Ravindra et al. 2008; Katsoyiannis et al. 2011).
Concentrations of PAHs were measured in different places worldwide. Some previously reported PAH concentrations indicated that they had a wide range depending on the sampling site. Greater PAH levels were generally measured in urban areas where many possible sources existed. Lower concentrations values were observed for greater-MW PAH species (>5 rings). This is because PAHs having greater MWs are mainly associated with particles and they can deposit at areas close to their sources, whereas PAHs in the gaseous phase can transport far away.
The behavior of PAHs in the atmosphere depends on complex physicochemical reactions, interactions with other pollutants, photochemical transformations, and dry and wet deposition. The transport, deposition, and chemical transformation of PAHs depend on their gas particle phase partitioning (Wang et al. 2011). The gas/particle partitioning of PAHs depends on the molecular weight of the compounds as well as temperature, humidity, and precipitation (Birgul et al. 2011).
Partitioning of the semivolatile organic compounds between the gas and particle phase has been studied thoroughly for the last years trying to predict atmospheric levels once the mechanism has been parameterized. Initially, partitioning was based on the Langmuir adsorption theory taking into account a mechanism of physical adsorption of the organic molecules on the particle surface. Afterwards, a mechanism based on the absorption of the semivolatile organic compounds in the organic phase of the particle matter was shown to be more relevant (Pankow 1994a, 1994b).
Finizio et al. (1997) described the partitioning of persistent organic pollutants between air and organic films on aerosols employing the octanol–air partition coefficient (K OA). Recently, a new theory was developed by taking into account PAH absorption in the organic matter as well as the adsorption on the particle matter soot (Dachs and Eisenreich 2000). The relative importance of both mechanisms in atmospheric aerosols has been evaluated through the comparison of the partition coefficients K OA and soot–air (K SA) in different urban atmospheres.
Ratios of individual PAHs can be employed as diagnostic tools to distinguish the sources of ambient PAHs (Park et al. 2002; Guo et al. 2003; Bourotte et al. 2005). For this purpose, some PAH ratios have been used to determine possible sources of PAHs (i.e., benz[a]anthracene [Ant] to benzo[a]pyrene [Pyr]). In addition to the diagnostic ratios, factor analysis was also introduced to determine the possible origins of PAHs in the ambient air. Some studies applied principal component analysis (PCA) for PAHs (Harrison et al. 1996; Simcik et al. 1999; Bourotte et al. 2005).
Bursa (40°10′58.17″N 29°4′6.32″E) is the fourth largest province in Turkey with a population approximately 2.5 million. Bursa is the center of automotive production; moreover, textile and fresh and canned food industries are present in the city’s organized industrial zones.
PAHs have been widely measured in different environmental sectors. However, limited studies are available on PAH pollution in the metropolitan Bursa. The present work investigated PAH concentrations, gas/particle partitioning, and seasonal and temporal variations at four different sampling sites in Bursa to understand the PAH source origins and their contributions.
Materials and Methods
Sampling Program and Collection
Ambient air samples were collected at four sampling sites: traffic, residential, coastal, and semirural. The traffic site samples were collected on a 2.5 m tall platform located in the garden of Tubitak Butal (TB [40°11′54.05″N-29°2′55.35″E]). The sampling site is surrounded by low-rise buildings, houses, and heavy traffic-loaded roads. This sampling site has been used in other studies (Cindoruk and Tasdemir 2010; Birgul et al. 2011). The residential sampling site, Yavuzselim (YS [40°11′48.40″N-29°5′46.80″E]) is located approximately 500 m away from the nearest major road. Ambient air samples were taken on the roof of a three-story building. The YS sampling site was surrounded by residences and small workplaces. In this region, natural gas and coal are mainly used as fuel. The semiurban samples were collected on a 1 m tall platform located on the roof of a four-story building at Gorukle Campus of the Uludag University Campus (UUC [40°13′40.66″N-28°52′35.11″E]), which is situated approximately 25 km away from the city center and nearly 1.5 km away from the nearest highway. This site is relatively far from any settlement zones and industrial facilities. The Mudanya (40°22′24.76″N-28°52′42.85″E) sampling site was chosen as a coastal area in the county of Mudanya. The sampling site was 700 m away from the Sea of Marmara, and the samples were collected from the top of a four-story building.
Thirty-five ambient air samples were collected between June 2008 and June 2009 from each sampling point at YS, TB and Mudanya, whereas 70 ambient air samples were collected from the UUC sampling point, yielding a total of 175 samples taken during the sampling campaign. Each month at least four samples were collected when there was no rain from each site. Each sampling duration was 24 h, but at the UUC site the duration was 2 × 12 h, i.e., two 12-h samples were collected at the UUC site per day. TSP samples were collected concurrently with the ambient samples. TSP values were determined by weighing the filter before and after sampling.
The air samples were collected using a high-volume sampler (HVAS) (Thermo Andersen GPS 11 Model, USA). To sample the particle and gas phases with the HVAS, a 10.2-cm glass fiber filter (GFF), two polyurethane foams (PUFs [5 cm in length]), and a cartridge (5 cm in diameter) were used in the sampler. The HVAS was calibrated using a standardized orifice manometer kit (Thermo Andersen) based on the manufacturer’s requirements for calibration.
The average air volumes for the PAHs were 261 ± 30, 287 ± 22, 191 ± 60, and 165 ± 62 m3 for the TB, YS, Mudanya, and UUC site, respectively. The flow rates were checked before and after sampling by calibrated flow meters. Field blanks were generated by equipping the sampler with a GFF and the PUFs without switching them on.
Before sampling, the GFFs were baked at 450 °C overnight. Then they were allowed to cool to room temperature in a desiccator. The PUF cartridges were Soxhlet-extracted with deionized (DI) water, methanol (MeOH), dichloromethane (DCM), and an acetone/hexane (ACE/HEX) (1:1) mixture for 24 h, dried in an oven at 60 °C, and stored in glass jars capped with Teflon-lined lids. After sampling, the GFFs and the PUF cartridges were stored at −20 °C in their containers (Cindoruk and Tasdemir 2010; Birgul et al. 2011).
Sample Preparation and Analysis
The full details of the sample preparation, analytical procedures, and quality assurance/quality control (QA/QC) have been given elsewhere (Cindoruk and Tasdemir 2010; Birgul et al. 2011; Birgul and Tasdemir 2011), but a brief summary is given here for completeness. All equipment used in the study was sequentially cleaned with hot tap water, DI water, MeOH, and ACE. To remove any contaminants, all of the tools were rinsed with DCM before use. All solvents and reagents used in the study were chromatographic-grade (Birgul and Tasdemir 2011; Birgul et al. 2011).
After sampling, the PUF cartridges were extracted for 24 h in the Soxhlet extractor using a DCM/petroleum ether (PE) (one-fourth) mixture. The GFFs were extracted with an ultrasonic bath (S 80 H Model; Elmasonic, Germany) with a 25-mL DCM/PE (1:4) mixture for 30 min. This step was repeated once.
Before extraction, all of the samples and blanks were spiked with the PAH surrogate standards. The certified PAH standard solutions (including the deuterium-labeled surrogate and the internal standards) were obtained from Dr. Ehrenstorfer GmbH (Augsburg, Germany). Five deuterated PAHs (naphthalene-d8 [Nap], acenaphthene-d10 [Ace], chrysene (Chr)-d12, perylene-d12, and phenanthrene-d10 [Phe]) were used as surrogate standard to monitor the analytical recovery efficiencies. Moreover, Pyr-d10 was used as the internal standard to determine the exact sample volume and was spiked into the extract just before injection.
The extract volumes were reduced and the solvent exchanged into hexane using a rotary evaporator and a high-purity N2 stream. After being concentrated to 2 mL, the samples were cleaned and fractionated on an alumina–silica column containing 3 g of silica, 2 g of alumina, and 2 g of Na2SO4 to separate the PAHs (Esen et al. 2006; Vardar et al. 2008; Birgul et al. 2011). The column was prewashed with 20 mL of DCM followed by 20 mL of PE. Then the sample in 2 mL of hexane was added to the column, and the PAHs were eluted with 20 mL of DCM. The final extracts were solvent exchanged into hexane and concentrated to 1 mL under a stream of N2. After the processes of extraction, volume reduction, and fractionation, the samples were ready for chromatographic analyses, and they were put in 2-mL vials and kept in a −20 °C freezer until analysis (Tasdemir and Esen 2007, 2008; Birgul and Tasdemir 2011).
All of the samples were analyzed on a gas chromatograph (Agilent 7890A) with a 30-m HP-5MS capillary column connected to an inert XL mass selective with a triple-axis mass-selective detector (Agilent 5975C) for 16 3- to 6-ring PAHs: Nap, acenaphthylene (Acy), Ace, fluorene (Flu), Phe, Ant, fluoranthene (Flt), Pyr, benz[a]anthracene (BaA), Chr, benzo[b]fluoranthene (BbF), benzo[k]fluoranthene (BkF), benzo[a]pyrene (BaP), dibenz[a,h]anthracene (DahA), benzo[g,h,i]perylene (BghiP), and indeno[1,2,3-cd]pyrene (IcdP). The details of the gas chromatography (GC)-mass spectrometry oven program, the sample preparation and the analytical method have recently been described (Birgul et al. 2011).
QA/QC
The average recoveries of the PAH surrogate standards are listed in Table 1. Some PAHs, such as Nap, Acy, Ace, and Flu, had lower recovery values (<50 %) and they were eliminated. The limit of detection (LOD) was determined as the mean blank mass plus three SDs (Simcik et al. 1998; Tasdemir et al. 2004; Tasdemir and Holsen 2005). The LODs for the PAHs ranged from 0.05 ng (DahA) to 64 ng (Phe). In general, the amounts of PAH in the samples were substantially greater than the amounts of LODs. The PAH quantities exceeding the LOD in the samples were quantified and blank corrected. The blank PUF cartridges and air filters were routinely placed in the field to determine whether there was any contamination during sampling, sample handling, or preparation. The concentration values were corrected with field blanks to eliminate the background contamination and artifacts by subtracting the average blank amount from the sample values (Simcik et al. 1998; Odabasi et al. 1999).
Results and Discussion
Ambient Air PAH Concentrations
The measured average total (gas + particle) ∑12PAH concentrations were 131 ± 160, 184 ± 276, 73 ± 108, and 27 ± 49 ng/m3 at the traffic (TB), residential (YS), coastal (Mudanya), and semiurban (UUC) site, respectively. The total concentration levels were highest at the YS site, followed by the TB site, the Mudanya site, and the UUC site.
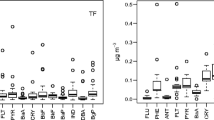
Seasonal average total concentration of PAHs for each site are shown in Fig. 1, whereas the variations of the total PAH concentration levels for each individual compound for the sampling sites are shown in Fig. 2.

Seasonal average total concentrations of PAHs for each site

Particle and gas phase PAH concentration levels for each individual PAH compound
All of the sampling points showed similar PAH compound variation trends and that PAHs were the most abundant compounds. The total PAH concentrations were dominated by the 2- to 4-ring PAHs, namely Phe, Flt and Pyr. The concentration of the greater molecular weight PAHs was low but preferentially enriched in particulates. The highest average total PAH concentration level was measured at the YS sampling site (184 ± 276 ng/m3), and it was approximately seven times greater than the lowest average concentration measured at the UUC sampling site (27 ± 49 ng/m3).
The values measured in the present study were within the range of previously reported values from other sites around the world (Table 2). A wide range of PAH levels have been reported in the literature. The existence of sources as well as the meteorological conditions can play an important role on the levels of PAHs. Some precautions have been effectively implemented to reduce the PAH concentration values in recent years, such as overlapping landfill areas, overlapping stabilization pools that were open to the atmosphere, sludge drying beds and controlling waste burning plants.
The YS sampling site was surrounded by residential areas and small workplaces, which mainly used natural gas and coal as fuel, and different types of materials such as plastic, cotton waste and wood were also used at the YS site for residential heating purposes. The TB sampling site had a heavy traffic load. Mudanya was the coastal sampling site, and the population density of the sampling site increased in the summer months. At this site, natural gas and coal were mainly used for residential heating. The UUC sampling site was relatively far away from sources, and the population density at this sampling site was lower than other sites. Various parameters such as the sampling site characteristics, the population density, the used fuel type, the meteorological conditions, and the removal and evaporation mechanisms can affect the obtained results.
In a study performed by our group at the TB and UUC sites between 2004 and 2005, similar PAHs were investigated, and the total (gas + particle) concentrations (∑12PAH) were calculated as 202 and 113 ng/m3, respectively (Tasdemir and Esen 2007; Vardar et al. 2008). The obtained results showed that the total PAH concentrations at the TB and UUC sampling sites have been decreasing over the years. The increase in natural gas use and strict control measures are thought to have affected the obtained results.
The ambient air PAH concentrations were analyzed during the heating and nonheating periods. A total of 12 samples were collected during the heating season (November to March), and 23 samples were collected during the nonheating season (April to October). The particle and gas phase PAH concentrations measured during the heating and nonheating periods are shown in Fig. 3. The average concentrations of Σ12PAHs in the heating period samples were approximately 5–7 times greater than those of the nonheating period samples. A similar seasonal trend, namely a greater concentration of PAHs in the winter, has been reported in several studies (Cincinelli et al. 2007; Terzi and Samara 2004; Fernandez et al. 2002).

Seasonal (nonheating versus heating seasons) variations of gas and particle phase PAH concentrations
In a study performed by our group at the UUC and the TB sampling sites between 2004 and 2005, PAHs were investigated during the heating and non heating periods (Esen et al. 2008b). During the heating period, average gas and particle PAH concentrations of 230.9 and 45.6 ng/m3 were found at UUC site, respectively. In contrast, during the nonheating period, average gas and particle PAH concentrations were 454,6 and 20.3 ng/m3, respectively, at the UUC site. In this study, the heating and nonheating period average PAH concentrations were determined to be 92 and 12 ng/m3 at the UUC, respectively. When the results were examined, the heating period PAH concentration levels tended to decrease over the years whereas the nonheating period PAH concentration values tended to increase. Decreased heating period PAH levels over time can closely be related to the changes in fuel type used for the heating purpose because the use of natural gas has increased during this period.
A seasonal fluctuation in the ambient air PAH concentrations were observed. The PAH levels within the sampling period generally showed greater concentrations in the colder season than in the warmer season. This difference is mainly caused by fossil fuel combustion for residential heating purposes (Park et al. 2002; Wu et al. 2005). The seasonal fluctuations in the gas and particulate phase PAH concentrations were shown in Fig. 3.
The lowest PAH concentrations were observed during the summer season for all of the sampling sites. The possible reasons for the lower PAH levels can be summarized as follows: lack of fuel burning for residential heating, reduced traffic load and no inversion layer formation. In addition, PAH compounds can be decomposed by means of photochemical reactions in the summer season.
Relationship with Meteorological Parameters
The effect of meteorological parameters (temperature, wind speed, wind direction and pressure) on the atmospheric concentrations of individual PAHs was investigated using multiple linear regression (MLR) analysis according to Eq. 1 (Hafner and Hites 2005).
where P is the partial pressure of the gas-phase PAHs (atm), T the average atmospheric temperature (K), U the wind speed (m/s) and WD the predominant wind direction (degrees). MLR was performed for the four sites using the same parameters, and the following equations were acquired:
Significant relationships were obtained between the meteorological data and the gas phase PAH concentrations for the YS and Mudanya sites, which have residential and coastal characteristics. The gas phase PAH concentration had a significant correlation (p < 0.05) with the wind direction in the YS and UUC sampling sites. In the TB and Mudanya sites, no significant relationship was found. All of the sampling sites had positive slopes for temperature, which shows that the PAH concentrations increase with decreasing air temperature. Different factors such as geographical characteristics, total suspended particle concentration, and the wind and the processes can affect obtained results. At the sampling sites, no significant relationship was found between the concentration-wind speed and the concentration-wind direction variables (p > 0.05).
The Clasius–Clapeyron equation (Eq. 2) can be used to determine the temperature dependence of the gas phase concentration:
According to Eq. 2, the ln P versus ln (1/T) relations were plotted, and the slope values for the four sites were determined. The slopes are 17351 2472 13686 and 9548 for the TB, YS, Mudanya and UUC sites, respectively.
Sitaras et al. (2004) obtained negative slope values (−4,650) for the ln P versus 1/T graph, whereas positive slope values were obtained in this study. Negative slope values indicated that the sources of the PAH are far from the sampling point, and positive slope values indicate that the PAH sources are close to the sampling point (Sofuoglu et al. 2001).
Similarly, Gaga et al. (2012) found positive slope values (5069.7) in their study, and they identified the effect of local sources on the gas phase concentrations of PAH compounds. Akyuz and Cabuk (2010) used the Clausius–Clapeyron equation and they also obtained positive slope values.
At the YS and UUC sites, a significant relationship (p < 0.05) was detected between the ambient air temperature and the gas phase concentration levels whereas at the other two sampling sites, no significant correlations were obtained between the temperature and the concentration levels (p > 0.05).
Gas/Particle Phase Partitioning
The gas and particle phase concentrations of individual PAHs at all of the sampling sites are listed in Table 3. For the traffic (TB), residential (YS), coastal (Mudanya) and semiurban (UUC) sites, the average gas phase total PAH (∑12PAH) concentrations were 132.64 ± 137.39 170.51 ± 232.46, 68.66 ± 81.62 and 21.47 ± 36.45 ng/m3, respectively, and the average particle phase total PAH (∑12PAH) concentrations were 29.86 ± 37.94, 57.39 ± 89.63 27.80 ± 43.22and 15.23 ± 26.63 ng/m3, respectively (Table 3). The gas/particle phase distributions indicated that approximately 81, 75, 71 and 59 % of the ∑12 PAHs were in the gas phase for the TB, YS, Mudanya and UUC sampling sites, respectively. Because of their greater volatility, the low to medium MW PAHs (to −5 rings) were more abundant in the gas phase, which is similar to the previous studies (Gevao et al. 1998; Fang et al. 2004a; Ohura et al. 2004; Tasdemir and Esen 2007).
The gas and particle-phase distributions of the PAH compounds in the ambient air samples were investigated, and the lower MW PAHs (Phe and Ant) were primarily present in the gas phase. Phe and Flt had the highest concentrations of all of the sampling sites. The highest gas and particle phase concentration values were obtained in the YS sampling site, whereas the lowest gas and particle phase concentration values were measured at the UUC sampling site.
The partition constant (K p [m3/μg]) normalized with the total suspended particle concentration (TSP [μg/m3]) is defined by Eq. (3) (Pankow 1987):
where C p and C g are the concentrations of PAHs in the particle and gas phases, respectively (ng/m3).
Two different mechanisms were used to explain the gas/particle PAH partitioning: physical adsorption onto the aerosol surface and absorption into the aerosol organic matter. Equation (4) relates K p with the subcooled liquid vapor pressure of the pure compound (P 0L , Pa or torr) (Pankow 1994a):
where mr and br are the empirical constants.
The ambient air temperature and the TSP concentration values of the collected samples ranged from 5.6 to 34 °C and from 13 to 379 μg/m3, respectively, in our studies. Figure 4 illustrates the relationship between log K p and log P 0L . Temperature dependent pL-values for PAHs were taken from Yamasaki et al. (1984).

Regression analysis of logK P versus log P 0L
Ideally, under equilibrium conditions, the slope mr should be equal to -1 for either adsorption or absorption mechanisms, explaining the gas/particle PAH partitioning (Pankow 1994a, b). It is remarkable that for adsorption, the differences between the enthalpies of the desorption and volatilization and the number of available adsorption sites must remain constant over a compound class. For absorption, the activity coefficients must remain constant over a compound class (Pankow 1987; Pankow and Bidleman 1992). Nevertheless, Goss and Schwarzenbach (1998) showed that deviations of m r = −1 are not always indicative of a lack of equilibrium. This deviation can be produced even if equilibrium sorption processes occur and sampling artifacts are negligible. Slopes, under certain conditions, could be indicative of the dominant mechanism, adsorption or absorption, determining gas/particle partitioning.
The values of the slopes for the collected samples were statistically significant (p < 0.05) in this work and varied between −0.52 and −1.84; the slopes are shallower than the expected slope under equilibrium conditions (−1). Similarly, the intercept of the relationship varied from −3.17 to −12.35. Slopes shallower than −0.6 are, in general, interpreted to be due to absorption into the organic matter, and values significantly steeper than −1 are due to the adsorption process (Goss and Schwarzenbach 1998).
The slope values obtained in this work varied widely. Therefore, it is not possible to positively identify the main gas–particle partitioning mechanism in the Bursa atmosphere, and the KP values were compared according to the two models in the next section. However, for the mr values obtained in this work, which were corroborated by other studies performed in other urban and rural areas, for which the slopes and intercepts of log K p versus log P 0L ranged from -0.43 to –1.04 and from –3.47 to –10.9, respectively (Tasdemir and Esen 2007; Simcik et al. 1998; Gustafson and Dickhut 1997; Baker and Eisenreich 1990).
Finizio et al. (1997) suggested that the octanol–air partition coefficient (K OA) can also be an alternate descriptor of gas/particle partitioning (Eq. 5) when the predominant process is absorption. K OA is the ratio of the concentration in octanol to the concentration in air when the octanol–air system is at equilibrium. This ratio can be employed as an interpretive value of the partitioning of semivolatile compounds between gas and particulate phases in the atmosphere (Vardar et al. 2004; Harner and Bidleman 1996, 1998; Finizio et al. 1997). Temperature dependent KOA values were calculated from Harner and Bidleman (1998):
where R is the universal gas constant (8.31 J/mol.K), T is the temperature (K), fOM is the fraction of organic matter on the particle, γ OCT and γ OM are the activity coefficients of the absorbing compound in octanol and the organic matter associated with the sampled particles, respectively, M OCT and M OM are the molecular weights of octanol (130 g/mol) and the organic matter, respectively, and ρ OCT is the density of octanol (820 kg/m3). An important portion of the organic matter may be secondary organic aerosol, which is formed by the oxidation of hydrocarbons and is most likely polar (Harner and Bidleman 1998; Pankow 1994a). γ OM may not be known and may vary among different compounds. If it is assumed that M OCT/M OM = 1 and γ OCT/γ OM = 1 (γ OM → 1 as X → 1 [Raoult’s law]), then the logarithmic form of Eq. 5 becomes:
There could be some problems associated with this approach when the slope of the Eq. (6) differs from unity (Tasdemir et al. 2004; Vardar et al. 2004; Harner and Bidleman 1998; Finizio et al. 1997). The possible reasons for this discrepancy are summarized as follows (Goss 2004; Goss and Schwarzenbach 1998, 2001): (1) adsorption onto some surfaces (i.e., minerals, salts and soot) is ignored; (2) the sorption characteristics of the organic parts in aerosols may differ from those of octanol, most likely because of numerous sources of the sampled particles; and (3) octanol may not be a representative compound for the species existing in aerosol.
Figure 5 shows the relationship between log K p and log K OA obtained from all of the sampling sites. The slopes of Eq. (6) ranged between 0.54 and 1.80 for the statistically significant cases (p < 0.05). Similarly, the intercept of the Eq. (6) ranged from −7.33 to −12.47. The slopes suggest that K OA is a good descriptor of the gas/particle partitioning process for the PAHs sampled in the scope of this study. In all cases, the r 2 values were high and ranged from 0.63 to 0.99. The obtained results are in line with the values reported previously for SVOCs. The slope and the intercept of log K p versus log K OA are 0.41–0.55 and –8.2 to –6.7, respectively, for PCBs and 0.55 and −8.23, respectively, for organochlorine compounds (Finizio et al. 1997; Lohmann et al. 2000). The plot of log K p versus log K OA for PAHs gives 0.79 and −10.01 for the slope and the intercept, respectively, according to Finizio et al. (1997), and these values are in good agreement with our values.

Relationship between logK p and log K OA
A soot/air partition coefficient (K SA) can be employed to investigate the K P values. In this approach, the PAHs are associated with soot particles. Elemental carbon (EC) was considered a surrogate for the soot phase (Dachs and Eisenreich 2000). Then, the modeled overall gas/particle partition coefficient, including the effects from both the absorption and the adsorption, is represented by Eq. 7 as follows:
where f EC is the fraction of the elemental carbon in the aerosol, a EC is the specific surface area of the elemental carbon and a AC is the surface area of the activated carbon. The a EC/a A ratio is assumed to be 1 (Dachs and Eisenreich 2000). f OM and f EC values of 0.15 and 0.05, respectively, were used to estimate K P in Eq. (7).
The temperature has an effect on the variables K OA and K SA, that is equal to K SW/H′, where K SW is the soot/water partition coefficient (Dachs and Eisenreich 2000) and H′ is the dimensionless Henry’s law constant (Poor et al. 2004). K SW is a function of temperature but only limited data are available. In contrast, the temperature dependence of K OA can be explained by the relationship presented below (Eq. 8):
where A and B were reported by Harner and Bidleman (1998).
Figures 6 and 7 show the relationship between the measured log K p and the modeled log K p(Oct) (Eq. 6) and between the measured log K p and the modeled log K p(Oct+Soot) (Eq. 7) obtained from this study. The K p values are examined statistically, and a significant correlation was found between the modelled and the experimental K p values (p < 0.001).

Relationship between measured log K p and modeled log K p(Oct)

Relationship between measured log K p and modeled log K p(Oct+Soot)
Compound Diagnostic Ratios for Source Identification
The molecular diagnostic ratios of some marker PAHs were used to identify the sources of PAHs in the environment (Guo et al. 2003; Katsoyiannis et al. 2011). PAHs are generally anthropogenic in origin and are major byproducts of the incomplete combustion of all types of organic materials (e.g., gasoline, diesel, and other fuels) (Fang et al. 2004b; Ravindra et al. 2008). PAH emission sources include the following: heavy oil combustion, natural gas combustion, wood and coal combustion, diesel combustion and vehicles (Yang and Chen 2004).
The molecular diagnostic ratios of PAH compounds were calculated for all sampling sites and the calculated values are listed in Table 4. The BaA/Chr ratio in Table 4 varied between 0.61 and 0.72, the BbF/BkF ratio varied between 1.28 and 1.8 for all of the sampling sites, and the high BbF/BkF and BaA/CHR ratios indicated the dominance of vehicle traffic over coal burning as a sources (Hwang and Foster 2006).
The BaP/BghiP ratio varied between 1.02 and 1.44 for all of the sampling sites: values <0.6 indicate the nontraffic emissions, and values >0.6 indicate the traffic emissions (Katsoyiannis et al. 2007). The calculated IcdP/BghiP ratios vary between 0.83 and 1.00 and indicate that coal burning was the main source for all of the sampling sites (Fang et al. 2004a).
The IcdP/IcdP +BghiP ratios vary between 1.18 and 5.18 for all sampling sites. Values <0.2 indicate the petrogenic sources; values between 0.2 and 0.5 indicate petroleum combustion; and values >0.5 indicate wood or coal combustion (Yunker et al. 2002). The BghiP/BaP ratio varied between 1.02 and 1.44 indicating diesel vehicle emissions (Manoli et al. 2004). Diagnostic ratios suggest traffic emission (gasoline + diesel), and minor coal and wood contributions. If you look at BbF/BkF ratio, you find that for the costal site, slight contribution of coal burning (ratio greater than the other sites).
In summary, the diagnostic PAH ratios suggested that traffic emissions (gasoline) and coal burning (residential heating), as well as other combustion processes, including wood burning, were the dominant PAH sources at all of the sampling sites.
Galarneau (2008) reported that it was not possible to positively identify the main sources of PAH compounds according to the diagnostic ratios because the calculated diagnostic ratios were very close to each other.
Source Estimations by PCA
PCA is a useful data analysis technique for examining factors and showing relationships and patterns within data sets. PCA has been widely used to identify the origins of organic compounds (Guo et al. 2003; Li et al. 2006). PCA results show which factors are able to explain the main part of the data variance (Fang et al. 2003).
PCA was performed using the SPSS statistical software packages (IBM SPSS 20). PCA was performed with varimax rotation, and principal components (factors) with eigenvalues > 1 were used to extract the possible sources. The factor analysis was performed for 12 PAHs.
The results from the application of PCA to our data indicated two main principal factors, which explained 97 % of the total variance in the data for particle phase concentration values of the TB. Factor 1, accounting for the majority of the total variance (56 %), was highly loaded with BghiP, Icdp, BkF, BaA, Chr, BbF, DahA, and BaP. Among these compounds, BkF, BaP and BghiP were considered to be the tracers of vehicle emissions (Duval and Friedlander 1981; Masclet et al. 1986; Miguel and Pereira 1989; Li and Kamens 1993; Harrison et al. 1996). The second factor, which accounted for 41 % of the total variance, was highly loaded with Flt, Pyr, BaA, BbF and DahA. These compounds are released into the atmosphere as a result of coal burning (Khalili et al. 1995; Simcik et al. 1999).
Three principal factors, explaining 99 % of the total variance in the data, were obtained by PCA at the TB sampling point for the gas phase PAHs. Factor 1, accounting for the majority of the total variance (43 %), was highly loaded with Ant, Chr, and BkF. These compounds are identified as being the source fingerprints of coal combustion and coke production (Larsen and Baker 2003). Therefore, factor 1 was selected to represent coal combustion. Factor 2, which accounted for 33 % of the total variance, was highly loaded with Flt, Pyr, BaA, BkF, and DahA, which are typical markers for pyrolysis or incomplete combustion (Wang et al. 2003). BkF is the component of fossil fuel (Rogge et al. 1993; Kavouras et al. 2001). Factor 3, constituting 23 % the total variance, was highly loaded with BkF and Chr.
Although the TB sampling point was considered a traffic site, the PCA results showed that coal combustion was another important PAH source there. The factor loadings of the PAH compounds at the TB sampling site are listed in Table 5.
Similarly, PCA was applied to the YS sampling point data. Two PCs were identified for the particle phase concentration values, and they explained 97 % of the total variance in the data. Factor 1, accounting for the majority of the total variance (53 %), was highly loaded with BaA, Chr, BbF, BkF, DahA, BaP, IcdP, and BghiP. Factor 2, which accounted for 44 % of the total variance, was highly loaded with Ant, Flt, Pyr, BaA, BbF, and DahA. Furthermore, three PCs were identified for the gas phase concentration values from the YS sampling point, and they explained 93 % of the total variance in the data. Factor 1, accounting for the majority of the total variance (35 %), was highly loaded with Phe, Ant, Flt, BaP, IcdP, and BghiP. The second factor accounted for 35 % of the total variance and was highly loaded with Ant, Flt, Pyr, BaA, BbF, and DahA. Factor 3 explained 23 % of the total variance and was highly loaded with Chr and BkF. The determined factor loadings of the PAHs at the YS sampling site (Table 5) indicated that combustion-based sources are dominant.
The PCA results showed that there were two PCs for the particle phase concentration values at the Mudanya sampling point, which accounted for 92 % of the total variance in the data. Factor 1, accounting for the majority of the total variance (64 %), was highly loaded with BaA, Chr, BbF, BkF, DahA, BaP, IcdP, and BghiP. Vehicle emissions were determined to be the main sources of BkF, BaP, and BghiP in the literature (Masclet et al. 1986; Miguel and Pereira 1989; Harrison et al. 1996). Factor 2, which accounted for 28 % of the total variance, was highly loaded with Phe, Ant, Flt, and Pyr. Flt was reported to be the dominant PAH in coke oven emissions (Khalili et al. 1995) and coal combustion (Masclet et al. 1987). Flt was often used as the tracer for coal combustion factor in PCA (Simcik et al. 1999), whereas Phe, Ant, and Pyr were identified as the source fingerprints of coal combustion (Duval and Friedlander 1981). For the gas phase, three PCs were identified at the Mudanya sampling point; these factors accounted for 100 % of the total variance in the data. Factor 1, accounting for the majority of the total variance (36 %), was highly loaded with Pyr, BaA, and IcdP. Factor 2 explained 34 % of the total variance and was highly loaded with Flt, BbF, BkF, and BghiP. Factor 3 accounted for 29 % of the total variance and was highly loaded with Chr, Phe, and Pyr (Table 5).
At the UUC sampling point, five PCs, accounting for 82 % of the total variance in the data, were identified for the particle phase concentration values. Factor 1, accounting for the majority of the total variance (29 %), was highly loaded with BaP, BghiP, IcdP, and Chr. The BaP and BghiP compounds indicated that vehicle emissions were more dominant (Miguel and Pereira 1989; Li and Kamens 1993). Factor 2, which accounted for 16 % of the total variance, was highly loaded with Flt, Pyr, and BbF. For the Pyr and BbF compounds, there was a negative correlation with PCA, and for Flt there was a positive correlation with the PCA. Factor 3 constituted of 13 % of the total variance and was highly loaded with BaP and BkF. A positive correlation was found between BaP and the PCA, whereas a negative correlation was found between BkF and the PCA. BkF is an indicator of diesel vehicle emissions (Li and Kamens 1993). Factor 4 accounted for the majority of the total variance (12 %), and was highly loaded with BaA and DahA. Factors 5 accounted for 11 % of the total variance and consisted of Pyr and BbF. Similarly, five PCs were identified for the gas phase at the UUC accounting for 97 % of the total variance in the data. Factor 1, accounting for the majority of the total variance (25 %), was highly loaded with BaP and DahA. Factor 2, accounting for 21 % of the total variance, was highly loaded with Phe and Flt. Factor 3, accounting for 19 % of the total variance, was highly loaded with BaA and Chr. Factor 4 explained 18 % of the total variance and consisted of Pyr and BkF, whereas factor 5 explained 14 % of the total variance and included only BbF (Table 5).
PCA results showed that pyrolytic sources affected the PAH concentrations at all of the sampling sites. Combustion sources, such as vehicles, incinerators and coal, are, as expected, generally negatively associated with temperature and wind velocity and positively correlated with relative humidity (Stelson and Seinfeld 1982). It is apparent from the results presented here that the use of PCA could be of practical significance in the qualification of pollution sources. Clearly separated, the 3-, 4- and 5-ring compounds enable the dominance of the pollution sources to the determined and their contamination origin to be predicted (O’Malley et al. 1996).
Conclusions
Ambient air PAH samples were collected at traffic, residential, coastal, and semiurban sites in Bursa, Turkey. Greater ambient PAH concentrations were measured in the gas-phase, and the ∑12PAH concentrations were dominated by lower MW PAHs. Seasonal variations in the PAH concentrations were investigated, and the concentration values were the highest for winter followed by the fall, spring, and summer. Ambient air PAH concentrations were analyzed during the heating and nonheating (n = 23) periods. The heating-period (fall and winter) concentration values were 5–7 times greater than the non-heating (spring and summer) period concentration values. MLR analysis indicated that the meteorological parameters were effective at the measured ambient PAH concentrations. The molecular diagnostic ratios showed that traffic emissions (gasoline and diesel) and coal-burning were the dominant PAH sources at all sampling sites. However, it was not possible to positively identify the main sources of the PAHs with these ratios because the calculated ratios were very close to each other. For this reason, PCA was used to identify the origins of the PAHs. The PCA results showed that the pyrolytic sources affected the PAH concentrations at all sampling sites.
References
Agency For Toxic Substances And Disease Registry (1994) Toxicological profile for polycyclic aromatic hydrocarbons (PAHs): Update. United States Department of Health and Human Services, Public Health Services, Atlanta
Akyuz M, Cabuk H (2010) Gas-particle partitioning and seasonal variation of polycyclic aromatic hydrocarbons in the atmosphere of Zonguldak, Turkey. Sci Total Environ 408:5550–5558
Baker JE, Eisenreich SJ (1990) Concentrations and fluxes of polycyclic aromatic-hydrocarbons and polychlorinated-biphenyls across the air-water interface of Lake Superior. Environ Sci Technol 24(3):342–352
Beegle LW, Wdowiak TJ, Harrison JG (2001) Hydrogenation of polycyclic aromatic hydrocarbons as a factor affecting the cosmic 6.2 micron emission band. Spectrochim Acta A 57:737–744
Birgul A, Tasdemir Y (2011) Seasonal atmospheric deposition variations of polychlorinated biphenyls (PCBs) and comparison of some deposition techniques. Environ Sci Pollut Res 18:396–406
Birgul A, Tasdemir Y, Cindoruk SS (2011) Atmospheric wet and dry deposition of polycyclic aromatic hydrocarbons (PAHs) determined using a modified sampler. Atmos Res 101:341–353
Bourotte C, Forti MC, Taniguchi S, Caruso M, Lotufo PA (2005) A wintertime study of PAHs in fine and coarse aerosols in Sao Paulo city, Brazil. Atmos Environ 39:3799–3811
Bozlaker A, Muezzinoglu A, Odabasi M (2008) Atmospheric concentrations, dry deposition and air–soil exchange of polycyclic aromatic hydrocarbons (PAHs) in an industrial region in Turkey. J Hazard Mater 153:1093–1102
Chirico R, Spezzano P, Cataldi D (2007) Gas-particle partitioning of polycyclic aromatic hydrocarbons during the spring and summer in a suburban site near major traffic arteries. Polycycl Aromat Compd 27:401–423
Cincinelli A, Del Bubba M, Martellini T, Gambaro A, Lepri L (2007) Gas-particle concentration and distribution of n-alkanes and polycyclic aromatic hydrocarbons in the atmosphere of Prato (Italy). Chemosphere 68:472–478
Cindoruk SS, Tasdemir Y (2010) Ambient air levels and trends of polychlorinated biphenyls (PCBs) at four different sites. Arch Environ Contam Toxicol 59(4):542–554
Dachs J, Eisenreich SJ (2000) Adsorption onto aerosol soot carbon dominates gas-particle partitioning of polycyclic aromatic hydrocarbons. Environ Sci Technol 34(17):3690–3697
Demircioglu E, Sofuoglu A, Odabasi M (2011) Atmospheric concentrations and phase partitioning of polycyclic aromatic hydrocarbons in İzmir, Turkey. Clean 39:319–327
Ding X, Wang XM, Xie ZQ, Xiang CH, Mai BX, Sun LG et al (2007) Atmospheric polycyclic aromatic hydrocarbons observed over the North Pacific Ocean and the Arctic area: spatial distribution and source identification. Atmos Environ 41:2061–2072
Duval MM, Friedlander SK (1981) Source resolution of polycyclic aromatic hydrocarbons in the Los Angeles atmosphere application of a CMB with first-order decay. United States EPA Report EPA-600/2- 81-161. United States Government Printing Office, Washington, DC
Esen F, Cindoruk SS, Tasdemir (2006) Ambient concentrations and gas/particle partitioning of polycyclic aromatic hydrocarbons in an urban site in Turkey. Environ Forensics 7:303–312
Esen F, Cindoruk SS, Tasdemir Y (2008a) Bulk deposition of polycyclic aromatic hydrocarbons (PAHs) in an industrial site of Turkey. Environ Pollut 152:461–467
Esen F, Tasdemir Y, Vardar N (2008b) Atmospheric concentrations of PAHs, their possible sources and gas-to-particle partitioning at a residential site of Bursa, Turkey. Atmos Res 88:243–255
Fang MD, Lee CL, Yu CS (2003) Distribution and source recognition of polycyclic aromatic hydrocarbons in the sediments of Hsin-ta Harbour and adjacent coastal areas, Taiwan. Mar Pollut Bull 46:941–953
Fang GC, Chang CN, Wu YS, Fu PPC, Yang IL, Chen MH (2004a) Characterization, identification of ambient air and road dust polycyclic aromatic hydrocarbons in central Taiwan, Taichung. Sci Total Environ 327:135–146
Fang GC, Chang KF, Lu C, Bai H (2004b) Estimation of PAHs dry deposition and BaP toxic equivalency factors (TEFs) study at urban, industry park and rural sampling sites in central Taiwan, Taichung. Chemosphere 55:787–796
Fernandez P, Grimalt JO, Vilanova RM (2002) Gas-particle partitioning of polycyclic aromatic hydrocarbons in high mountain regions of Europe. Environ Sci Technol 36:1162–1168
Finizio A, Mackay D, Bidleman T, Harner T (1997) Octanol-air partitioning coefficients as a predictor of partitioning of semi-volatile organic chemicals to aerosols. Atmos Environ 31(15):2289–2296
Gaga EO, Ari A, Döğeroğlu T, Çakırca EE, Machin NE (2012) Atmospheric polycyclic aromatic hydrocarbons in an industrialized city, Kocaeli, Turkey: study of seasonal variations, influence of meteorological parameters and health risk estimation. J Environ Monit 14:2219–2229
Galarneau E (2008) Review: source specificity and atmospheric processing of airborne PAHs: implications for source apportionment. Atmos Environ 42:8139–8149
Garivait H, Polprasert HC, Yoshizumi K, Baetz Reutergardh L (2001) Airborne polycyclic aromatic hydrocarbons (PAH) in Bangkok urban air: Part II Level and distribution. Polycycl Aromat Compd 18:325–350
Gevao B, Jones KC, Hamilton-Taylor J (1998) Polycyclic aromatic hydrocarbon (PAH) deposition to and processing in a small rural lake, Cumbria, UK. Sci Total Environ 215:231–242
Goss KU (2004) The air/surface adsorption equilibrium of organic compounds under ambient conditions. Crit Rev Environ Sci Technol 34:339–389
Goss KU, Schwarzenbach RP (1998) Gas/solid and gas/liquid partitioning of organic compounds: critical evaluation of the interpretation of equilibrium constants. Environ Sci Technol 32:2025–2032
Goss KU, Schwarzenbach RP (2001) Linear free energy relationships used to evaluate equilibrium partitioning of organic compounds. Environ Sci Technol 35:1–9
Guo H, Lee SC, Ho KF, Wang XM, Zou SC (2003) Particle-associated polycyclic aromatic hydrocarbons in urban air of Hong Kong. Atmos Environ 37(38):5307–5317
Gustafson K, Dickhut R (1997) Particle/gas concentrations and distributions of PAHs in the atmosphere of southern Chesapeake Bay. Environ Sci Technol 31:140–147
Hafner WD, Hites RA (2005) Effects of wind and air trajectory directions on atmospheric concentrations of persistent organic pollutants near the Great Lakes. Environ Sci Technol 39:7817–7825
Halasinski TM, Salama F, Allamandola LJ (2005) Investigation of the ultraviolet, visible, and near-infrared absorption spectra of hydrogenated polycyclic aromatic hydrocarbons and their cations. Astrophys J 628:555–566
Hanedar A, Alp K, Kaynak B, Baek J, Avsar E, Odman MT (2011) Concentrations and sources of PAHs at three stations in Istanbul, Turkey. Atmos Res 99:391–399
Harner T, Bidleman TF (1996) Measurements of octanol-air partition coefficients for polychlorinated biphenyls. J Chem Eng Data 41:895–899
Harner T, Bidleman TF (1998) Octanol-air partition coefficient for describing particle/gas partitioning of aromatic compounds in urban air. Environ Sci Technol 32:1494–1502
Harrison RM, Smith DJT, Luhana L (1996) Source apportionment of atmospheric polycyclic aromatic hydrocarbons collected from an urban location in Birmingham, UK. Environ Sci Technol 30:825–832
Hwang HM, Foster GD (2006) Characterization of polycyclic aromatic hydrocarbons in urban storm water runoff flowing into the tidal Anacostia River. Environ Pollut 140:416–426
Inomata Y, Kajino M, Sato K, Ohara T, Kurokawa JI, Ueda H et al (2012) Emission and atmospheric transport of particulate PAHs in northeast Asia. Environ Sci Technol 46:4941–4949
International Agency for Research on Cancer (1983) Polynuclear aromatic compounds, Part 1. Chemical environmental and experimental data. IARC monographs on the evaluation of the carcinogenic risk of chemicals to humans. Volume 32. IARC, Lyon, France, pp 33–91
Katsoyiannis A, Terzi E, Cai QY (2007) On the use of PAH molecular diagnostic ratios in sewage sludge for the understanding of the PAH sources. Is this use appropriate? Chemosphere 69(8):1337–1339
Katsoyiannis A, Sweetman AJ, Jones KC (2011) PAH molecular diagnostic ratios applied to atmospheric sources: a critical evaluation using two decades of source inventory and air concentration data from the UK. Environ Sci Technol 45:8897–8906
Kavouras IG, Koutrakis P, Tsapakis M, Lagoudaki E, Stephanou EG, Baer DV et al (2001) Source apportionment of urban particulate aliphatic and polynuclear aromatic hydrocarbons (PAHs) using multivariate methods. Environ Sci Technol 35:2288–2294
Khalili NR, Scheff PA, Holsen TM (1995) PAH source fingerprints for coke ovens, diesel and gasoline engines, highway tunnels, and wood combustion emissions. Atmos Environ 4:533–542
Kishida MMC, Fujimori K, Imamura K, Takenaka N, Maeda Y, Lan TTN et al (2009) Seasonal change in the atmospheric concentration of particulate polycyclic aromatic hydrocarbons in Ho Chi Minh City, Vietnam. Bull Environ Contam Toxicol 83:747–751
Larsen RK, Baker JE (2003) Source apportionment of polycyclic aromatic hydrocarbons in the urban atmosphere: a comparison of three methods. Environ Sci Technol 37(9):1873–1881
Lee RGM, Jones KC (1999) The influence of meteorology and air masses on daily atmospheric PCB and PAH concentrations at an UK location. Environ Sci Technol 33:705–712
Li CK, Kamens RM (1993) The use of polycyclic aromatic hydrocarbons as sources signatures in receptor modeling. Atmos Environ A 27:523–532
Li J, Zhang G, Li XD, Qi SH, Liu GQ, Peng XZ (2006) Source seasonality of polycyclic aromatic hydrocarbons (PAHs) in a subtropical city, Guangzhou, South China. Sci Total Environ 355:145–155
Lim MCH, Ayoko G, Morawska L (2005) Characterization of elemental and polycyclic aromatic hydrocarbon compositions of urban air in Brisbane. Atmos Environ 39:463–476
Liu G, Tong Y, Luong JHT, Zhang H, Sun H (2010) A source study of atmospheric polycyclic aromatic hydrocarbons in Shenzhen, South China. Environ Monit Assess 163:599–606
Lohmann R, Harner T, Thomas GO, Jones KC (2000) A comparative study of the gas-particle partitioning of PCDD/Fs, PCBs, and PAHs. Environ Sci Technol 34:4943–4951
Mandalakis M, Tsapakis M, Tsoga A, Stephanou EG (2002) Gas-particle concentrations and distribution of aliphatic hydrocarbons, PAHs, PCBs and PCDD/Fs in the atmosphere of Athens (Greece). Atmos Environ 36:4023–4035
Manoli E, Kouras A, Samara C (2004) Profile analysis of ambient and source emitted particle-bound polycyclic aromatic hydrocarbons from three sites in Northern Greece. Chemosphere 56:867–878
Martellini T, Giannoni M, Lepri L, Katsoyiannis A, Cincinelli A (2012) One year intensive PM2.5 bound polycyclic aromatic hydrocarbons monitoring in the area of Tuscany, Italy. Concentrations, source understanding and implications. Environ Pollut 164:252–258
Masclet P, Mouvier G, Nikolaou K (1986) Relative decay index and sources of polycyclic aromatic hydrocarbons. Atmos Environ 20:439–446
Masclet P, Bresson MA, Mouvier G (1987) Polycyclic aromatic hydrocarbons emitted by power stations, and influence of combustion conditions. Fuel 66:556–562
Mastral AM, Callen MS (2000) A review on polycyclic aromatic hydrocarbon (PAH) emissions from energy generation. Environ Sci Technol 34:3051–3057
Miguel AH, Pereira PAP (1989) Benzo(k)fluoranthene, benzo(ghi)perylene, and indeno(1,2,3-cd)pyrene: news tracers of automotive emissions in receptor modeling. Aerosol Sci Technol 10:292–295
Morville S, Delhomme O, Millet M (2011) Seasonal and diurnal variations of atmospheric PAH concentrations between rural, suburban and urban areas. Atmos Pollut Res 2:366–373
Odabasi M, Vardar N, Sofuoglu A, Tasdemir Y, Holsen TM (1999) Polycyclic aromatic hydrocarbons (PAHs) in Chicago air. Sci Total Environ 227:57–67
Ohura T, Amagai T, Fusaya M, Matsushita H (2004) Spatial distributions and profiles of atmospheric polyaromatic hydrocarbons in two industrial cities in Japan. Environ Sci Technol 38:49–55
O’Malley V, Abrajano T, Hellou J (1996) Stable carbon isotopic apportionment of individual polycyclic aromatic hydrocarbons in St John’s Harbour, Newfoundland. Environ Sci Technol 30:634–639
Ozcan S, Aydin ME (2009) Polycyclic aromatic hydrocarbons, polychlorinated biphenyls and organochlorine pesticides in urban air of Konya, Turkey. Atmos Res 93:715–722
Pankow JF (1987) Review and comparative analysis of the theories on partitioning between the gas and aerosol particulate phases in the atmosphere. Atmos Environ 21:2275–2283
Pankow JF (1994a) An absorption model of gas/particle partitioning of organic compounds in the atmosphere. Atmos Environ 28:185–188
Pankow JF (1994b) An absorption model of gas/aerosol partitioning involved in the formation of secondary organic aerosol. Atmos Environ 28:189–193
Pankow JF, Bidleman TF (1992) Interdependence of the slopes and intercepts from log-log correlations of measured gas-particle partitioning and vapor pressure—I. Theory and analysis of available data. Atmos Environ A 26:1071–1080
Park SS, Kim YJ, Kang CH (2002) Atmospheric polycyclic aromatic hydrocarbons in Seoul, Korea. Atmos Environ 36:2917–2924
Poor N, Tremblay R, Kay H, Bhethanabotla V, Swartz E, Luther M et al (2004) Atmospheric concentrations and dry deposition rates of polycyclic aromatic hydrocarbons (PAHs) for Tampa Bay, Florida, USA. Atmos Environ 38:6005–6015
Possanzini M, Di Palo V, Gigliucci P, Tomasi Sciano MC, Cecinato A (2004) Determination of phase-distributed PAH in Rome ambient air by denuder/GC-MS method. Atmos Environ 38:1727–1734
Rajput N, Lakhani A (2010) Measurements of polycyclic aromatic hydrocarbons in an urban atmosphere of Agra, India. Atmosfera 23:165–183
Ravindra K, Mittal AK, Van Grieken R (2001) Health risk assessment of urban suspended particulate matter with special reference to polycyclic aromatic hydrocarbons: a review. Rev Environ Health 16:169–189
Ravindra K, Bencs L, Wauters E, de Hoog J, Deutsch F, Roekens E et al (2006) Seasonal and site-specific variation in vapour and aerosol phase PAHs over Flanders (Belgium) and their relation with anthropogenic activities. Atmos Environ 40:771–785
Ravindra K, Sokhia R, Grieken RV (2008) Atmospheric polycyclic aromatic hydrocarbons: source attribution, emission factors and regulation. Atmos Environ 42:2895–2921
Rogge WF, Hildemman LM, Mazukek MA, Cass GR, Simoneit BRT (1993) Sources of fine organical aerosol. 5. Natural gas home appliances. Environ Sci Technol 27(13):2736–2744
Rubailo AI, Oberenko AV (2008) Polycyclic aromatic hydrocarbons as priority pollutants. J Siberian Fed Univ Chem 4(1):344–354
Simcik MF, Franz TP, Zhang H, Eisenreich SJ (1998) Gas-particle partitioning of PCBs and PAHs in the Chicago urban and adjacent coastal atmosphere: states of equilibrium. Environ Sci Technol 32(2):251–257
Simcik MF, Eisenreich SJ, Lioy PJ (1999) Source apportionment and source/sink relationships of PAHs in coastal atmosphere of Chicago and Lake Michigan. Atmos Environ 33:5071–5079
Sitaras LE, Bakeas EB, Siskos PA (2004) Gas/particle partitioning of seven volatile polycyclic aromatic hydrocarbons in a heavy traffic urban area. Sci Total Environ 327:249–264
Sofuoglu A, Odabasi M, Tasdemir Y, Khalili NR, Holsen TM (2001) Temperature dependence of gas-phase polycyclic aromatic hydrocarbon and organochlorine pesticide concentrations in Chicago air. Atmos Environ 35:6503–6510
Stelson AW, Seinfeld JH (1982) Relative humidity and temperature dependence of the ammonium nitrate dissociation constant. Atmos Environ 16:983–992
Tang N, Hattori T, Taga R, Igarashi K, Yang X, Tamura K, Kakimoto H (2005) Polycyclic aromatic hydrocarbons and nitropolycyclic aromatic hydrocarbons in urban air particulates and their relationship to emission sources in the Pan-Japan Sea Countries. Atmos Environ 39:5817–5826
Tasdemir Y, Esen F (2007) Urban air PAHs: concentrations, temporal changes and gas/particle partitioning at a traffic site in Turkey. Atmos Res 84:1–12
Tasdemir Y, Esen F (2008) Deposition of polycyclic aromatic hydrocarbons (PAHs) and their mass transfer coefficients determined at a trafficked site. Arch Environ Contam Toxicol 55(2):191–198
Tasdemir Y, Holsen TM (2005) Measurement of particle phase dry deposition fluxes of polychlorinated biphenyls (PCBs) with a water surface sampler. Atmos Environ 39:1845–1854
Tasdemir Y, Vardar N, Odabasi M, Holsen TM (2004) Concentrations and gas/particle partitioning of PCBs in Chicago. Environ Pollut 131:35–44
Terzi E, Samara C (2004) Gas-particle partitioning of polycyclic aromatic hydrocarbons in urban, adjacent coastal and continental background sites of western Greece. Environ Sci Technol 38:4973–4978
Tsapakis M, Stephanou EG (2005) Occurrence of gaseous and particulate polycyclic aromatic hydrocarbons in the urban atmosphere: study of sources and ambient temperature effect on the gas/particle concentration and distribution. Environ Pollut 133:147–156
Vardar N, Tasdemir Y, Odabasi M, Noll KE (2004) Characterization of atmospheric concentrations and partitioning of PAHs in the Chicago atmosphere. Sci Total Environ 327:163–174
Vardar N, Esen F, Tasdemir Y (2008) Seasonal concentrations and partitioning of PAHs in a suburban site of Bursa, Turkey. Environ Pollut 155:298–307
Wang D, Xu X, Chu S, Zhang D (2003) Analysis and structure prediction of chlorinated polycyclic aromatic hydrocarbons released from combustion of polyvinylchloride. Chemosphere 53:495–503
Wang XY, Li QB, Luo YM, Ding Q, Xi LM, Ma JM et al (2010) Characteristics and sources of atmospheric polycyclic aromatic hydrocarbons (PAHs) in Shanghai, China. Environ Monit Assess 165:295–305
Wang X, Westerdahl D, Wu Y, Pan X, Zhang KM (2011) On road emission factor distributions of individual diesel vehicles in and around Beijing, China. Atmos Environ 45:503–513
Wing MR, Bada JL (1992) The origin of polycyclic aromatic hydrocarbons in meteorites. Orig Life Evol Biosph 21:375–383
World Health Organization Regional Office for Europe (1987) Polynuclear aromatic hydrocarbons (PAH). Air quality guidelines for Europe. WHO, Copenhagen, pp 105–117
Wu SP, Tao S, Xu FL, Dawson R, Lan T, Li BG et al (2005) Polycyclic aromatic hydrocarbons in dustfall in Tianjin, China. Sci Total Environ 345:115–126
Yamasaki H, Kuwata K, Kuge Y (1984) Determination of vapor-pressure of polycyclic aromatic-hydrocarbons in the supercooled liquid-phase and their adsorption on airborne particulate matter. Nippon Kagaku Kaishi 101(8):1324–1329 (in Japanese)
Yang HH, Chen CM (2004) Emission inventory and sources of polycyclic aromatic hydrocarbons in the atmosphere at a suburban area in Taiwan. Chemosphere 56:879–887
Yunker MB, McDonald RW, Vingarzan R, Mitchell RH, Goyotte D, Sylvestre S (2002) PAHs in the Fraser River basin: a critical appraisal of PAH ratios as indicators of PAH source and composition. Org Geochem 33:489–515
Acknowledgments
This study was financially supported by The Scientific and Technological Research Council of Turkey (Project No. 107Y165).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Birgul, A., Tasdemir, Y. Concentrations, Gas-Particle Partitioning, and Seasonal Variations of Polycyclic Aromatic Hydrocarbons at Four Sites in Turkey. Arch Environ Contam Toxicol 68, 46–63 (2015). https://doi.org/10.1007/s00244-014-0105-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00244-014-0105-8