
Abstract
The combined effects of lead (Pb) and cadmium (Cd) on primary cultures of rat proximal tubular (rPT) cells were studied. These cells were either treated with Pb acetate (0.5 and 1 μM) alone, Cd acetate (2.5 and 5 μM) alone, or a combination of Pb and Cd acetate, and then joint cytotoxicity was evaluated. The results showed that the combination of these two metals decreased cell viability and increased the number of apoptotic and necrotic cells and lactate dehydrogenase release synergistically. Simultaneously, increased intracellular reactive oxygen species, malondialdehyde, and calcium levels and decreased mitochondrial membrane potential, intracellular acidification, and inhibition of Na+, K+-, and Ca2+-ATPase activities were shown during the exposure. In addition, apoptotic morphological changes induced by these treatments in rPT cells were demonstrated by Hoechst 33258 staining. The apoptosis was markedly prevented by N-acetyl-l-cysteine, whereas necrosis was not affected. In summary, there was a synergistic cytototic effect of Pb combined with Cd on rPT cells. Cell death induced by Pb–Cd mixture was mediated by an apoptotic and a necrotic mechanism. Apoptotic death was the chief mechanism. Changes of intracellular events were intimately correlated with both oxidative stress and mitochondrial dysfunction, which promoted the development of apoptosis.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Lead (Pb) and cadmium (Cd) are known occupational hazards and environmental contaminants having long biological half-lives and multiorgan toxicity. Unlike complex organic pollutants, Pb and Cd compounds cannot be degraded by microorganisms; instead, they can be accumulated by organisms and also take part in the process of bioaccumulation throughout the food chain, thus threatening human health (Kong et al. 1995). As a consequence of increasing industrialization, large quantities of Pb and Cd are discharged into the environment, and the levels of these metals have increased substantially during the past several years (Nriagu and Pacyna 1988; Peñuelas and Filella 2002; Liu 2003). In reality, Pb and Cd are often released simultaneously in the environment from a number of natural and anthropogenic sources, adverse health effects caused by combined exposure to Pb and Cd have provoked significant public health concerns (Liu 2003; Brender et al. 2006; Walker et al. 2007).
The kidney is a sensitive target organ for Pb or Cd exposure (Nolan and Shaikh 1992; Włostowski et al. 2008). The nephrotoxicity induced by Pb and/or Cd has been extensively reported in many literatures (Ercal et al. 1996; Loghman-Adham 1997; Patra et al. 2001; Antonio Garcia and Corredor 2004; Morales et al. 2006; Muntner et al. 2007; Wang et al. 2009a). Most of the these reports dealt with a single Pb or Cd exposure. A few studies have been conducted to investigate the combined effects of Pb and Cd on renal injury. Moreover, most previous experiments concentrated on animal and cell line studies, and primary cultures of tubular cells were minimally applied (Wang et al. 2009b). We recently demonstrated that synergism exists between the two metals in their nephrotoxic effects in in vivo studies (Wang et al. 2009c, 2010). Cell cultures are valuable tools for mechanistic studies at the cellular level. However, little information is available about the combined effect of Pb and Cd on nephrotoxicity in vitro. Therefore, the objective of this study was to set up an in vitro system to investigate the joint effects of Pb and Cd on nephrotoxicity. Due to the fact that primary cultures can better represent live tissue than cultures of permanent cell lines, they are ideal for in vitro toxicity studies. Moreover, the renal tubule is one major site of renal impairment (Goyer 1989); thus, a pure preparation of isolated proximal tubular cells from rat kidneys was used for culture in this study. Our previous studies confirmed that apoptotic cell death mediated by oxidative stress plays a chief role in renal damage induced by a single Pb or Cd exposure in vitro at lower exposure levels (Wang et al. 2009b, d). Hereby, a series of indices related to oxidative stress and apoptosis were chosen to elucidate the joint cytotoxicity of Pb and Cd in primary cultures of rat proximal tubular (rPT) cells.
Materials and Methods
Chemicals
All chemicals were of highest grade purity available. Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Laboratories (Tokyo, Japan) and Annexin V-FITC Apoptosis Detection kit from Pharmingen (Becton Dickinson, USA). E-cadherin (L-CAM) was purchased from BD Transduction Laboratories (Lexington, KY). Fetal bovine serum (FBS) was purchased from Sijiqing Biological Engineering Material (Hangzhou, China). Malondialdehyde (MDA) assay kit, Na+- and K+-ATPase assay kit, and Ca2+-ATPase analysis kit were from Keygen Biotech (Nanjing, Jiangsu, China). N,N,N′,N′-tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN), Fluo-3/AM, and 2′,7′-bis (2-carboxyethyl)-5,6-carboxyfluorescein/acetoxymethyl ester (BCECF/AM) were purchased from Molecular Probes, USA. DMEM-F12 (1:1), Pb acetate (PbAc2), Cd acetate (CdAc2), propidium iodide (PI), 2′,7′-dichlorofluorescein diacetate (DCFH-DA), Hoechst 33258 staining, antibiotic-antimycotic solution, rhodamine 123 (Rh-123), collagenase, trypsin, N-acetyl-l-cysteine (NAC), ethylene diamine tetraacetic acid (EDTA) and all other chemicals were purchased from Sigma-Aldrich, USA.
Cell Isolation, Cell Culture, and Exposure Conditions
Isolation of rPT cells was described in our previous study (Wang et al. 2009b). The primary cells reached confluence after 144 h of cultivation and were subcultured after a 6-day culture by the trypsin-EDTA digestive method. Both primary cells and subcultures were cultured in DMEM/F12 medium supplemented with 25 mM HEPES, 10% FBS (heat-inactivated at 56°C), 0.25 g/L glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in the presence of 95% air and 5% carbon dioxide (CO2). Identity of the proximal tubular cells was confirmed by staining with antibodies against specific proximal tubular antigens (immunocytochemical staining). Staining with E-cadherin (L-CAM) antibody was performed to confirm absence of other kidney cell types, such as distal tubular, collecting duct, and glomerulum-derived cells (Nouwen et al. 1993). Cell purity of the isolated proximal tubular cells was routinely >95%. The first passage was used to perform the experimental design when it (the first-passage culture) was cultured for 120 h, at which point there was the highest cell viability (according to the growth curve [data not shown]). Based on the doses of Pb or Cd in previous studies (Wang et al. 2009b, d), cell cultures undergoing exponential growth were incubated at 37°C for 12 h in the presence of 0 and 2.5 μM Cd (C1) and 5 μM Cd (C2) combined with or without Pb acetate at 0 and 0.5 μM (P1) and 1 μM (P2) in a serum-free medium. Primarily, events of the low-dose combined group (P1C1) and high-dose combined group (P2C2), i.e., 0.5 μM Pb + 2.5 μM Cd and 1 μM Pb + 5 μM Cd, were chosen for specific study.
Cell Viability Assay
CCK-8 is a one-bottle solution that contains water-soluble tetrazolium salt. It can reduce dehydrogenase in the mitochondria to water-soluble formazan dyes. The absorbance of these formazan dyes at 450 nm is proportional to the number of viable cells in the medium. Cells were seeded at a density of 1 × 104 in 96-well plates. After preprocessing, the cells were treated with a series of Pb and/or Cd concentrations (0 and 0.5 μM Pb, 1 μM Pb, 2.5 μM Cd, 5 μM Cd, 0.5 μM Pb + 2.5 μM Cd, 0.5 μM Pb + 5 μM Cd, 1 μM Pb + 2.5 μM Cd, and 1 μM Pb + 5 μM Cd) for a time period of 3, 6, 12, and 24 h. Also, the cells were incubated with two combined concentrations of Pb + Cd (0.5 μM Pb + 2.5 μM Cd and 1 μM Pb + 5 μM Cd) and 100 μM NAC for 12 h to assess the cytoprotective effect of NAC on cell survival. At the destined time points, cell viability assays were performed using CCK-8 according to manufacturer’s instructions. The absorbance was read at 450 nm by the microplate reader (Sunrise, Austria).
Lactate Dehydrogenase Release
Lactate dehydrogenase (LDH) activity was measured as the rate of decrease of the absorbance at 340 nm resulting from the oxidation of NADH to NAD (Koh and Choi 1987). For the determination of LDH, the culture medium was collected after 12 h of heavy-metal exposure, and the adherent cells on the plates were washed with phosphate buffered saline (PBS) and then lysed with 0.1 M Tris-HCl (pH 7.4) containing 0.1% Triton X-100. LDH activity was measured both in culture medium and in cell lysate after the addition of 1 mM pyruvate and 0.2 mM β-NADH. LDH release was given as percentage of LDH in the culture medium with respect to total LDH (the sum between LDH in the culture medium and LDH inside the cells).
Hoechst 33258 Staining
Apoptotic morphological changes in the nuclear chromatin were detected by staining with the DNA binding fluorochrome Hoechst 33258 (bisbenzimide). rPT cells were seeded on sterile cover glasses placed in the 24-well plates. After incubation with various concentrations of heavy metal (0, P1, P2, C1, C2, P1C1, and P2C2) for 12 h, cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min and then incubated with Hoechst 33258 staining solution (50 μM in PBS) for 10 min in the dark at room temperature. After three washes with PBS, the cells were viewed under an inverted fluorescence microscope (IX-70; Olympus, Japan) at an excitation wavelength of 352 nm through fluorescein isothiocyanate (FITC) filter (blue fluorescence). For assessing the extent of the apoptosis induced by heavy-metal treatments, 200 cells were randomly selected to count those apoptotic cells within every batch of experiment, and each one was performed in triplicate.
Flow Cytometric Analysis
All the following assays were carried out on BD-FACS Aria flow cytometer, and the 488 nm laser was utilized. Cells were seeded at a density of 3 × 105 in six-well plates and treated with various Pb and/or Cd concentrations (0, P1, P2, C1, C2, P1C1, and P2C2) for 12 h. In addition, 100 μM NAC was coadministered with P1C1/P2C2 for 12 h to assess its effect on apoptosis. After treatment, the adherent cells were collected with trypsin-EDTA solution (1.25 g/L trypsin and 0.2 g/L EDTA). The detached and adherent cells were pooled and harvested by centrifugation at 1500 rpm for 5 min. The harvested cells (at least 1 × 106 cells/group) were washed twice with PBS and incubated with various fluorescent dyes for flow cytometric analysis. Cell debris, characterized by a low forward scatter/side scatter (FSC/SSC) value, was excluded from analysis. The data were analyzed by Cell Quest software, and mean fluorescence intensity was obtained by histogram statistics.
Assessment of Apoptotic and Necrotic Cells
Detection of apoptotic and necrotic cell distribution was performed using annexin V/PI staining assay. The amount of early apoptotic cells, late apoptotic cells, living cells, and necrotic cells was determined as the percentage of annexin V+/PI−, annexin V+/PI+, annexin V−/PI−, and annexin V−/PI+ cells, respectively (Vermes et al. 1995). Therefore, the total apoptotic proportion included the percentage of cells with fluorescence annexin V+/PI− and annexin V+/PI+. After the exposure (12 h), the harvested cells were labeled with annexin V-FITC and PI using an apoptosis-detection kit according to the manufacturer’s protocol. FITC and PI fluorescence was measured through FL-1 filter (530 nm) and FL-2 filter (585 nm), respectively, and 10,000 cells were analyzed.
Mitochondrial Membrane Potential (Δψ) Assay
For the detection of mitochondrial ΔΨ, the harvested cells were incubated with rhodium-123 (5 μg/mL final concentration) for 30 min in the dark at 37°C, harvested, and resuspended in PBS. Mitochondrial ΔΨ was measured by the fluorescence intensity (FL-1, 530 nm) of 10,000 cells.
Reactive Oxygen Species Measurement
Generation of reactive oxygen species (ROS) was monitored by measurement of hydrogen peroxide generation using the fluorescent probe dichlorofluorescein (DCFH)-DA. This dye is cleaved to form nonfluorescent dichlorofluorescein (DCFH) in the cells, which is oxidized to fluorescent DCF by ROS. Thus, the DCF fluorescence intensity is proportional to the amount of peroxides produced by the cells. Briefly, 1.5 × 106 harvested cells/mL were incubated with DCFH-DA (100 μM final concentration) for 30 min in the dark at 37°C. The incubated cells were harvested and suspended in PBS, and ROS generation was measured by the fluorescence intensity (FL-1, 530 nm) of 10,000 cells on the flow cytometer.
Analysis of Intracellular Free Ca2+ Concentration
After treatment, the harvested cells were incubated with 0.5 mmol/L TPEN (cell-permeant heavy-metal chelator that has high affinity for Cd2+ or Pb2+ and low affinity for Ca2+) to discount Pb and/or Cd interference in the fluorescence signal because calcium and heavy metals (Pb and Cd) interact similarly (Dyatlov et al. 1998; Thévenod 2009). Cells were loaded with Fluo-3/AM (1 μM final concentration) for 30 min in the dark at 37°C and then washed with D-Hank’s balanced salt solution (Ca2+- and Mg2+-free). Intracellular [Ca2+]i levels were represented with fluorescent intensity (FL-1, 530 nm) of 10,000 cells on the flow cytometer.
Measurement of Intracellular pH with BCECF
Intracellular pH (pHi) was measured by loading cells with membrane-impermeant dye BCECF. The detailed procedure was performed according to Hirpara et al. (2001), so the process will only be mentioned briefly. The harvested cells were incubated with BCECF/AM (2 μM final concentration) in 1 mL serum-free culture medium. After 30 min of incubation in the CO2 incubator at 37°C, cells were pelleted, rinsed twice with PBS buffer, and resuspended at an appropriate density for fluorescence measurements. A minimum of 10,000 events were analyzed, and the ratio of BCECF fluorescence at 525 and 610 nm was used to obtain pHi from a calibration curve.
Determination of Other Indices
Cells were seeded at a density of 6 to approximately 8 × 105 in four-well plates and treated with the heavy metal (0, P1, P2, C1, C2, P1C1, and P2C2) for 12 h. After treatment, the detached and adherent cells were pooled and harvested by centrifugation to measure the following assays.
Activities of Na+-, K+-, and Ca2+-ATPase
The harvested cells were homogenized in ice-cold physiological saline in an ultrasonic disintegrator. The cell homogenates were centrifuged at 1000×g for 10 min, and supernatants were obtained whose protein concentration was determined by the Folin phenol method using bovine serum albumin as a standard (the protein concentration in the assay was 3 to approximately 5 mg/mL) (Lowry et al. 1951). ATPase activities were assayed by the quantization of phosphonium ions, which was performed in accordance with the ATPase-detection protocol.
Measurement of Intracellular Lipid Peroxides
The harvested cells were lysed in ice-cold physiological saline by sonication followed by centrifugation at 15,000×g for 5 min at 4°C. The resulting supernatants were used immediately for measuring the levels of MDA. The quantification was based on measuring formation of thiobarbituric acid (TBA)-reactive substances according to the manufacturer’s protocol. TBA was added to each sample tube and vortexed. The reaction mixture was incubated at 95°C for 40 min. After cooling, the chromogen was read spectrophotometrically at 532 nm. Protein concentrations of the samples were determined according to the method of Lowry et al. (1951) to normalize the levels of MDA.
Statistical Analysis
Data are presented as means ± SDs from at least three independent experiments with different batches of cells, each one performed in duplicate or triplicate. Intergroup differences were assessed by one-way analysis of variance (ANOVA) after ascertaining the homogeneity of variance between the treatments. To assess the individual effects of Pb and Cd on cell viability at different times, a factorial experiment design consisting of three concentrations of Pb and Cd was performed and followed by two-way ANOVA. Synergistic effect was evaluated when the interaction between Pb and Cd was significant (p < 0.05). All statistical analyses were carried out using SPSS 13.0 (SPSS, Chicago, IL, USA [IL: import license]). The critical value for statistical significance was p < 0.05.
Results
Effects of Pb and/or Cd on Cell Viability
As listed in Table 1, either Pb or Cd alone caused progressive cell death in rPT cells in a concentration- and time-dependent fashion during the exposure. Whereas 1 μM Pb and 5 μM Cd effectively affected cell viability after a 3-h incubation period (p < 0.05), all of the single treatments significantly decreased cell viability after a 6-h exposure time (p < 0.05). By one-way ANOVA, the cell viabilities of the four combined treatments (P1C1 [0.5 μM Pb + 2.5 μM Cd], P1C2 [0.5 μM Pb + 5 μM Cd], P2C1 [1 μM Pb + 2.5 μM Cd], and P2C2 [1 μM Pb + 5 μM Cd]) were significantly decreased compared with the control after a 3-h exposure time (p < 0.01). Moreover, cell death caused by the combined treatments (P1C1, P1C2, P2C1, and P2C2) was always higher than that by the corresponding Pb treatment or Cd treatment at the same exposure time, respectively. Furthermore, cell viabilities resulting from P2C2 (1 μM Pb + 5 μM Cd) were always lower than those of any other treatment at the destined time points (3, 6, 12, and 24 h). Two-way ANOVA indicated that Cd alone had a significant inhibitory effect on cell viability after a 3-h exposure time (p < 0.05), whereas Pb alone caused significant progressive cell death after a 6-h incubation period (p < 0.05). In addition, both Pb and Cd exhibited a strong synergistic interaction on cell death after the beginning of incubation for ≥6 h (p < 0.05).
Protective Effect of NAC on Cell Death Induced by Heavy-Metal Exposure
Cytoprotective actions of NAC on cell death induced by single Pb and single Cd treatment were investigated in our previous studies (Wang et al. 2009b, d). Hereby, NAC was chosen to evaluate its protective effect on cell death induced by the combination of Pb and Cd exposure. As shown in Fig. 1, the cell death induced by Pb + Cd (P1C1 and P2C2) was prevented in part by 100 μM NAC (p < 0.05). In addition, NAC treatment alone did not affect cell viability (p > 0.05).

Cytoprotective action of NAC on cell death induced by heavy-metal exposure as measured by CCK-8 reduction method. The cells were incubated with heavy metal (P1C1, P2C2) and/or 100 μM NAC for 12 h to determine cell survival. Cont control, P1C1 0.5 μM Pb + 2.5 μM Cd, and P2C2 1 μM Pb + 5 μM Cd. Two different colors were chosen to indicate which cells were treated with NAC (+) and which were not (−); i.e., white columns for no NAC treatment and black for NAC treatment. Data are presented as means ± SDs (n = 6). * Statistical significance between control and heavy-metal treatments (P1C1 and P2C2) among the no NAC-treated groups; # statistical significance between cells cultured in the absence (−) and presence (+) of NAC. NS not significant. ** p < 0.01; # p < 0.05
Effects of Pb and Cd Compounds on Necrosis and Apoptosis
Cell death induced by Pb combined with Cd was performed by the necrotic mechanism judged by the LDH release (Fig. 2a). After 12 h of exposure, the necrosis induced by Pb + Cd (P1C1 and P2C2) increased significantly (p < 0.01). However, necrosis was not inhibited by the antioxidant molecule (NAC) (p > 0.05). In addition, necrosis induced by combined treatments of Pb and Cd was reflected by flow cytometric analysis (Fig. 2b). The number of necrotic cells in the two combined groups was significant higher than that of the control group, respectively. In any case, necrosis was not inhibited by NAC (p > 0.05). Apoptosis induced by Pb combined with Cd was performed by flow cytometric analysis (Fig. 2b). At 12 h, the number of apoptotic cells enhanced significantly (p < 0.01) to 9.33-fold and 14.3-fold of that of the control after exposure to P1C1 and P2C2, respectively; apoptosis was significantly prevented by NAC (p < 0.05). However, NAC treatment alone did not affect necrosis and apoptosis (p > 0.05). In addition, apoptosis and necrosis induced by Pb and Cd, singly and in combination, are shown in Figure 3. The number of apoptotic cells and necrotic cells in the P1C1 and P2C2 treatments were significantly (p < 0.05) increased as compared with the corresponding single Pb treatment or Cd exposure, respectively. These results indicate that Pb combined with Cd induced apoptosis and necrosis in rPT cells, which exhibited a synergistic effect.

The combined effects of Pb and Cd on cell death type. Effect of NAC (1) on necrosis (a) and b as measured by LDH release and flow cytometry and (2) on apoptosis (b) as measured by flow cytometry. Cont control, P1C1 0.5 μM Pb + 2.5 μM Cd, and P2C2 1 μM Pb + 5 μM Cd. The cells were incubated with two metal compounds (P1C1 and P2C2) and/or 100 μM NAC for 12 h to measure the assays. Two different colors were chosen to point out which cells were treated with NAC (+) and which were not (−); i.e., white columns for no NAC treatment and black for NAC treatment. Data are presented as means ± SDs of three separate experiments, and each one was performed in triplicate. * Statistical significance between control and heavy-metal treatments (P1C1 and P2C2) among the no NAC-treated groups; # statistical significance between cells cultured in the absence (−) and presence (+) of NAC. NS not significant. ** p < 0.01; # p < 0.05

Effects of Pb and Cd, singly and in combination, on apoptosis and necrosis in rPT cells after 12 h of exposure. Cont control, P1 0.5 μM Pb, P2 1 μM Pb, C1 2.5 μM Cd, C2 5 μM Cd, P1C1 0.5 μM Pb + 2.5 μM Cd, and P2C2 1 μM Pb + 5 μM Cd. Results are means ± SDs of three separate experiments, and each one was performed in triplicate. * Statistical significance between control and heavy-metal treatments (P1, P2, C1, C2, P1C1, and P2C2); # statistical significance between the combined groups (P1C1 and P2C2) and the single Pb (P1 and P2) or Cd (C1 and C2) groups, respectively. ** p < 0.01; # p < 0.05
Effects of Pb and Cd Compounds on Apoptotic Morphological Changes
Apoptotic morphological changes in the nuclear chromatin were detected by staining with the DNA binding fluorochrome Hoechst 33258 (bisbenzimide) (Fig. 4a). In the control group, the majority of cells had uniformly stained nuclei, and the chromatin of normal nuclei was unaltered and spread uniformly throughout the entire nucleus. After exposure to Pb and/or Cd for 12 h, it showed morphological changes typical of apoptosis, i.e., fragmented chromatin was characterized by a scattered, drop-like structure, and condensed chromatin was located at the periphery of the nuclear membrane and appeared as a half-moon form. The nuclei of apoptotic cells appeared smaller and shrunken compared with intact cells. In addition, the statistical result (Fig. 4b) of cells with apoptotic characteristics shows that the number of apoptotic cells enhanced progressively with Pb or Cd concentration when exposed to each metal alone. As far as the extent of the apoptosis is concerned, the combined groups caused more severe lesions than did the respective single Pb or Cd exposure (p < 0.05). Furthermore, the number of apoptotic cells resulting from Pb (1 μM) + Cd (5 μM) combination was much greater than those of any other treatment.

Effects of Pb and/or Cd on apoptotic morphological changes in rPT cells as determined by Hoechst 33258 staining. a Cont control, P1 0.5 μM Pb, P2 1 μM Pb, C1 2.5 μM Cd, C2 5 μM Cd, P1C1 0.5 μM Pb + 2.5 μM Cd, and P2C2 1 μM Pb + 5 μM Cd. In the heavy-metal exposed groups, changes of nuclei fragmentation with condensed chromatin are evident (thin arrows). Cont-P2C2 = original magnification ×400. b The statistical results of apoptotic cells are expressed as means ± SDs of three separate experiments, and each one was performed in triplicate. Cont control, P1 0.5 μM Pb, P2 1 μM Pb, C1 2.5 μM Cd, C2 5 μM Cd, P1C1 0.5 μM Pb + 2.5 μM Cd, and P2C2 1 μM Pb + 5 μM Cd. * Statistical significance between control and heavy-metal treatments (P1, P2, C1, C2, P1C1, and P2C2); # statistical significance between the combined groups (P1C1 and P2C2) and the single Pb (P1 and P2) or Cd (C1 and C2) groups, respectively. ** p < 0.01; # p < 0.05
Effects of Pb and Cd Compounds on ROS Generation and Intracellular MDA Levels
The generation of intracellular ROS was detected by using the fluorescent probe DCFH-DA. As shown in Fig. 5a, the level of ROS was increased by treatment with single Pb or Cd alone in a dose-dependent manner. Exposure to the combined metals led to a greater increase in ROS generation compared with the single heavy-metal exposure from 3.81-fold to 4.53-fold that of the control after exposure to Pb (0.5 μM) + Cd (2.5 μM) and Pb (1 μM) + Cd (5 μM), respectively. Simultaneously, the level of intracellular MDA (a common end product of lipid peroxidation) was measured to study the impact of heavy-metal exposure on oxidative lesions (Fig. 5b). The concentration-dependent generation of MDA levels increased significantly (p < 0.01) after a single Pb or Cd exposure, and the 0.5 μM Pb, 1 μM Pb, 2.5 μM Cd, and 5 μM Cd treatments exhibited a 3.5-, 4.5-, 3.1-, and 4.6-fold significant (p < 0.01) increase, respectively, compared with the control group. Also, levels of MDA in the combined treatments (P1C1 and P2C2) were significantly (p < 0.05) higher than those of the single Pb (P1 and P2) or single Cd (C1 and C2) treatments, respectively.

Effects of Pb and/or Cd on generation of ROS (a) and intracellular MDA levels (b). a The harvested cells were incubated with 100 μM DCFH-DA for 30 min at 37°C. DCF fluorescence was measured using flow cytometer with FL-1 filter. Fluorescence results were expressed as mean fluorescence intensity. Each bar represents mean ± SD (n = 6). b The cells were incubated with different treatments of Pb and/or Cd for 12 h to measure MDA levels. Data are presented as means ± SDs of three experiments from cells of different cultures, and each one was performed in triplicate. Cont control; P1 0.5 μM Pb, P2 1 μM Pb, C1 2.5 μM Cd, C2 5 μM Cd, P1C1 0.5 μM Pb + 2.5 μM Cd, and P2C2 1 μM Pb + 5 μM Cd. * Statistical significance between control and heavy-metal treatments (P1, P2, C1, C2, P1C1, and P2C2); # statistical significance between the combined groups (P1C1 and P2C2) and the single Pb (P1 and P2) or Cd (C1 and C2) groups, respectively. ** p < 0.01; # p < 0.05
Effects of Pb and Cd Compounds on Mitochondrial Membrane Potential (ΔΨ)
Rhodium-123, a lipophilic cationic fluorescent dye, is selectively taken up by mitochondria and used to analyze changes in mitochondrial ΔΨ during apoptotic cell death. As shown in Fig. 6, a significant decrease in mitochondrial ΔΨ occurred in rPT cells after exposure to Pb and Cd for 12 h, both singly and in combination. Regarding the single-metal treatments, its reduction occurred in a concentration-dependent manner. Likewise, both Pb and Cd exhibited a significant synergistic effect on the loss in mitochondrial ΔΨ (p < 0.01).

Effects of Pb and/or Cd on mitochondrial membrane potential. rPT cells were treated with Pb and Cd (singly and in combination) for 12 h. Rhodium-123 was added, and the harvested cells were incubated for 30 min. Fluorescence was measured using flow cytometer with FL-1 filter. Fluorescence results were expressed as mean fluorescence intensity. Each bar represents mean ± SD (n = 6). Cont control, P1 0.5 μM Pb, P2 1 μM Pb, C1 2.5 μM Cd, C2 5 μM Cd, P1C1 0.5 μM Pb + 2.5 μM Cd, and P2C2 1 μM Pb + 5 μM Cd. * Statistical significance between control and heavy-metal treatments (P1, P2, C1, C2, P1C1, and P2C2); # statistical significance between the combined groups (P1C1 and P2C2) and the single Pb (P1 and P2) or Cd (C1 and C2) groups, respectively. ** p < 0.01; # p < 0.05
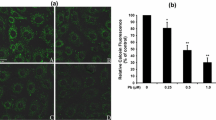
Effects of Pb and Cd Compounds on Intracellular [Ca2+]i and Activities of Na+-, K+-, and Ca2+-ATPase
After incubation with PbAc2 (0.5 and 1 μM) and/or CdAc2 (2.5 and 5 μM) for 12 h, marked enhancement of intracellular [Ca2+]i (p < 0.01) and significant inhibition of Na+- and K+-ATPase activities and Ca2+-ATPase activity (p < 0.05, p < 0.01) occurred (Fig. 7). Intracellular calcium overload and loss in activities of ATPases (Na+, K+, and Ca2+) induced by either Pb or Cd alone occurred in a concentration-dependent manner. The combined Pb and Cd treatments had a significant synergistic effect on intracellular [Ca2+]i, being 2.4-fold and 3.0-fold of that of the control value after exposure to Pb (0.5 μM) + Cd (2.5 μM) and Pb (1 μM) + Cd (5 μM), respectively. Simultaneously, the Pb–Cd mixture had a significant inhibitory effect on the activities of ATPases, levels far lower than those of the single-metal treatments. When exposed to Pb (0.5 μM) + Cd (2.5 μM) and Pb (1 μM) + Cd (5 μM) for 12 h, Na+- and K+-ATPase activities decreased to 58.2% and 46.7% of the control value and Ca2+-ATPase activity to 64.4% and 53.1% of the control value, respectively.

Effects of Pb and/or Cd on intracellular [Ca2+]i levels (a) and the ATPase activities (b and c) in rPT cells. a The collected cells were first incubated with 0.5 mmol/L TPEN, and then the harvested cells were incubated with 1 μM Fluo-3/AM for 30 min at 37°C. Fluo-3 fluorescence was measured using flow cytometer with FL-1 filter. Fluorescence results were expressed as mean fluorescence intensity. Each bar represents mean ± SD (n = 6). The cells were treated for 12 h with the indicated heavy-metal treatments to measure the activities of Na+- and K+-ATPase (b) and Ca2+-ATPase (c). Data are presented as mean ± SD of three experiments from cells of different cultures, and each one was performed in triplicate. Among the groups Cont control, P1 0.5 μM Pb, P2 1 μM Pb, C1 2.5 μM Cd, C2 5 μM Cd, P1C1 0.5 μM Pb + 2.5 μM Cd, and P2C2 1 μM Pb + 5 μM Cd. * Statistical significance between control and heavy metal treatments (P1, P2, C1, C2, P1C1, and P2C2); # statistical significance between the combined groups (P1C1 and P2C2) and the single Pb (P1 and P2) or Cd (C1 and C2) groups, respectively. * p < 0.05; ** p < 0.01; # p < 0.05
Intracellular Acidification Due to the Induction of Pb and/or Cd
The results of intracellular pH analysis by flow cytometry are shown in Fig. 8. A significant decrease in pHi occurred in rPT cells exposed to Pb and/or Cd, which indicates that low-dose heavy metal treatments resulted in intracellular acidification in rPT cells. The decrease of pHi in the rPT cells exposed to either Pb or Cd alone occurred in a dose-dependent manner. The result of pHi in Pb (1 μM) + Cd (5 μM) treatment was significantly (p < 0.05) decreased compared with the 1 μM Pb treatment or 5 μM Cd treatment, respectively; its level was far lower than those of other treatments.

Effects of Pb and/or Cd on intracellular pH of rPT cells as measured by flow cytometry. The harvested cells were loaded with 2 μM BCECF/AM for 30 min at 37°C, and the fluorescence ratio (525/610 nm) obtained was used to determine pHi. Each bar represents mean ± SD (n = 6). Cont control, P1 0.5 μM Pb, P2 1 μM Pb, C1 2.5 μM Cd, C2 5 μM Cd, P1C1 0.5 μM Pb + 2.5 μM Cd, and P2C2 1 μM Pb + 5 μM Cd. * Statistical significance between control and heavy-metal treatments (P1, P2, C1, C2, P1C1, and P2C2); # statistical significance between the combined groups (P1C1 and P2C2) and the single Pb (P1 and P2) or Cd (C1 and C2) groups, respectively. * p < 0.05; ** p < 0.01; # p < 0.05
Discussion
Cd and Pb are known nephrotoxic toxicants. With regard to the present studies, little is known about joint effects of these metals on nephrotoxicity, although they are commonly found associated in polluted environments. Furthermore, it remains controversial as to the joint effects of Pb and Cd on nephrotoxicity (Antonio Garcia and Corredor 2004; Wang et al. 2009c, 2010). Due to the fact that primary cultures can better represent live tissue, they are ideal for in vitro toxicity studies. Moreover, the proximal tubule is a sensitive target site of renal damage induced by Cd and Pb exposure (Goyer 1989). Herein, this study was designed to investigate the intracellular events in primary cultures of rat proximal tubular cells to aid in better understanding of the underlying nature of nephrotoxic damage by coexposure to Pb and Cd. Primarily, events of heavy-metal exposure during a 12-h period were chosen to investigate the toxic effects.
Concerning the single-metal exposures, the progressive loss in cell viability shows that Cd or Pb exposure induced cell death in these cells, depending on both the metal concentration and the exposure time (Table 1). Moreover, the combined groups caused more severe cell death than the corresponding single Pb or Cd treatment at the same exposure time, respectively. Cell death induced by the combined groups was correlated with both the incubation time and the mixed concentrations of Pb and Cd. Factorial-design ANOVA showed that these metals exhibited a significant interaction on cell death after the beginning of incubation ≤ 6 h (p < 0.05). Thus, there was an obvious synergistic effect of Pb combined with Cd on cytotoxicity in rPT cells. To conveniently investigate the cytotoxic mechanism, only low-dose combination (0.5 μM Pb + 2.5 μM Cd) and high-dose combination (1 μM Pb + 5 μM Cd) treatments were chosen for specific study. Our previous studies demonstrated that single Pb- or Cd-induced cytotoxicity is intimately related to oxidative stress (Wang et al. 2009b, d). As shown in Fig. 1, cell death was in part prevented by NAC, an antioxidant agent, indicating that oxidative stress could be implicated in the mechanism by which Pb combined with Cd induces cell death in rPT cells. Meanwhile, cell death induced by Pb–Cd mixture is mediated by two mechanisms: necrotic and apoptotic (Fig. 2). The increased LDH release rates (Fig. 2a) and number of necrotic cells (Fig. 2b) seen in the combined treatments were remarkable, indicating cytosolic membrane breaking. It could be demonstrated that a necrotic mechanism played a role in cell death. In addition, a significant increase in the number of apoptotic cells induced by Pb–Cd mixture was implicated with apoptotic death (Fig. 2b). Compared with apoptotic cells, the number of necrotic cells was small. In other words, apoptotic death played a primary role in cell death induced by Pb–Cd mixture at these combined concentrations. Apoptosis induced by Pb–Cd mixture was significantly prevented by NAC (Fig. 2b) whereas necrosis was not affected by this antioxidant molecule (Fig. 2a, b). It also appears that in apoptotic death mediated by Pb combined with Cd, but not in necrotic death, oxidative stress could be implicated. Thus, this study showed a certain type of relationship between apoptosis and oxidative stress in the joint effects of Pb and Cd on rPT cells. Necrotic death induced by these combinations is not discussed here but is worthy of further investigation.
Regarding apoptotic rates (Fig. 3), Pb–Cd mixtures caused the more severe apoptotic effect than did each metal alone, and the number of apoptotic cells resulting from Pb (1 μM) + Cd (5 μM) combination treatment was much greater than those of any other treatments. Also, apoptosis induced by these metals was assessed by Hoechst 33258 staining (Fig. 4). Compared with intact cells, obvious apoptotic morphological changes were mediated by Pb and Cd exposure, both singly and combined (Fig. 4a). The percentage of apoptotic cells (Fig. 4b) was significantly increased after exposure to Pb and/or Cd for 12 h, which is consistent with the result obtained by flow cytometric analysis (Fig. 3). Apoptotic morphological changes further show that Pb–Cd combination exhibited an obvious synergistic interaction on apoptosis in rPT cells.
In addition, the ability of either Pb or Cd alone to induce oxidative stress in rPT cells was confirmed in our previous studies (Wang et al. 2009b, d). In this study, cells treated with different combinations of Pb and Cd generated ROS significantly (Fig. 5a) accompanied by the enhanced generation of intracellular MDA levels (Fig. 5b), thus demonstrating further that oxidative stress plays a critical role in cytotoxicity induced by Pb–Cd combination. These changes also indicated that Pb and Cd exhibit a strong synergistic effect on lipid peroxidation in rPT cells during treatment. Mitochondria are the major source of intracellular ROS generation as well as an important target for the damaging effects of ROS (Ott et al. 2007). Lipid peroxidation is involved in mitochondrial dysfunction, and alterations in mitochondrial function are largely attributed to the damaging effects of ROS (Sen et al. 2006). Moreover, excessive amounts of ROS in the mitochondria can directly oxidize and inactivate proteins, leading to further ROS overload (Franco et al. 2009). In this study, the induction of ROS in rPT cells could be mediated by mitochondria alterations because both Pb and Cd, singly and in combination, produced a breakdown of mitochondrial ΔΨ (Fig. 6). The combined effect of Pb and Cd on mitochondrial dysfunction is consistent with the results in vivo studies (Wang et al. 2010). ROS has been demonstrated to perform certain functions in early stages of apoptosis and to induce depolarization of the mitochondrial membrane, which also indicates that ROS may constitute a direct cause of mitochondrial dysfunction (Oh and Lim 2006). A variety of investigations have demonstrated that ROS is a prerequisite for the collapse of mitochondrial ΔΨ (Bolduc et al. 2004; Yang et al. 1997). In single Pb or Cd exposure, it has been shown that the mitochondria are most pertinent in mediating apoptosis in rPT cells, putatively by way of metal-induced ROS (Wang et al. 2009b, d). Because accumulation of ROS targeted the mitochondrial membrane to induce a collapse of mitochondrial ΔΨ, it is reasonable to assume that mitochondria play a crucial role in apoptosis induced by Pb combined with Cd.
Furthermore, previous studies have suggested that intracellular calcium overload is associated with apoptosis induced by either Pb or Cd alone in rPT cells (Wang et al. 2009b, d). In this study, we observed that treatment with Pb–Cd combination also resulted in abnormal manifestation in intracellular [Ca2+]I by showing that significantly stronger [Ca2+]i existed in the combined groups than in single-metal treatment, respectively (Fig. 7a). This fact suggests that abnormal Ca2+ homeostasis due to combined exposure to Pb and Cd may be another important mechanism of the development of apoptosis in these cells. Intracellular Ca2+ levels are maintained by many factors. Among them, Na+-, K+-, and Ca2+-ATPase play critical roles in intracellular Ca2+ homeostasis, which act in the removal of Ca2+ from the cytosol across the plasma membrane, and blockage of these enzymes can induce cytosolic calcium overload (Fujita et al. 1998). Inhibition of Na+-, K+-, and Ca2+-ATPase activities by Pb and/or Cd exposure in rPT cells (Fig. 7b, c) may demonstrate this idea. The Na+-, K+-, and Ca2+-ATPases are concerned with immediate release of energy, and sufficient ATP is necessary to maintain their function (Üner et al. 2005). Because mitochondria are the major site of ATP production and mitochondrial ΔΨ is the driving force of ATP synthesis, a breakdown in mitochondrial ΔΨ could lead to a decrease in ATP levels (Chakraborti et al. 1999). Therefore, the resultant deficiency in cellular ATP levels due to mitochondrial dysfunction leads to inhibition of the Na+-, K+-, and Ca2+-ATPases (Wang et al. 2003; Qin et al. 2008). Simultaneously, Na+-, K+-, and Ca2+-ATPases are susceptible to the effects of oxidative damage (Qin et al. 2008). Thus, it can be concluded that mitochondrial dysfunction induced by Pb combined with Cd, leading to the disruption of energy metabolism and overproduction of ROS (elevation of intracellular MDA levels), results in decreased ATPase activities, which are involved in intracellular calcium overload.
In addition to its role in the modulation of cytosolic calcium levels, mitochondria may act as a mediator in the equilibrium of intracellular pH effects during the process of apoptosis (Foster et al. 2006). Cytosolic pH changes have been reported to be involved in mitochondria-mediated apoptosis, with cytosolic acidification promoting cytochrome c-mediated activation of caspases (Matsuyama et al. 2000). Moreover, it has been reported that oxidative stress rapidly decreases pHi in kidney epithelial cell lines (Kaufman et al. 1993). As shown in Fig. 8, intracellular acidification was accompanied by exposure of Pb and/or Cd to rPT cells. Likewise, there was an obvious synergistic effect of Pb combined with Cd on the decrease of pHi in rPT cells. Combined with the changes in the above-mentioned indices, it can be seen that intracellular acidification in rPT cells results from the induction of oxidative stress, which could create an intracellular environment efficient for caspase activation to promote apoptosis (Hirpara et al. 2001). Furthermore, intracellular acidification was related to the mitochondrial function because changes in cytosolic pH may directly affect the ΔΨ to make it negative (Foster et al. 2006). This will further result in the dysfunction of mitochondria.
In summary, oxidative stress plays a critical role in cell death induced by Pb combined with Cd in rPT cells. Apoptosis triggered by oxidative stress was the chief death mechanism in these cells. The death mechanism induced by Pb–Cd mixture was similar to that in cells exposed to single Pb or Cd at these concentrations, resulting from a synergistic interaction in the combination of these metals. ROS-related alteration of mitochondrial homeostasis (breakdown of mitochondrial ΔΨ and ROS overproduction), as well as disturbance in the equilibrium of intracellular milieu (intracellular acidification and calcium overload), indicate their crucial roles as intracellular mediators for the initiation of apoptotic events.
References
Antonio Garcia T, Corredor L (2004) Biochemical changes in the kidneys after perinatal intoxication with lead and/or cadmium and their antagonistic effects when coadministered. Ecotoxicol Environ Saf 57:184–189
Bolduc JS, Denizeau F, Jumarie C (2004) Cadmium-induced mitochondrial membrane-potential dissipation does not necessarily require cytosolic oxidative stress: studies using rhodamine-123 fluorescence unquenching. Toxicol Sci 77:299–306
Brender JD, Suarez L, Felkner M, Gilani Z, Stinchcomb D, Moody K et al (2006) Maternal exposure to arsenic, cadmium, lead, and mercury and neural tube defects in offspring. Environ Res 101:132–139
Chakraborti T, Das S, Mondal M, Roychoudhury S, Chakraborti S (1999) Oxidant, mitochondria and calcium: an overview. Cell Signal 11:77–85
Dyatlov VA, Dyatlova OM, Parsons PJ, Lawrence DA, Carpenter DO (1998) Lipopolysaccharide and interleukin-6 enhance lead entry into cerebellar neurons: application of a new and sensitive flow cytometric technique to measure intracellular lead and calcium concentrations. Neurotoxicology 19:293–302
Ercal N, Treeratphan P, Hammond TC, Matthews RH, Grannemann N, Spitz D (1996) In vivo indices of oxidative stress in lead exposed C57BL/6 mice are reduced by treatment with meso-2,3-dimercaptosuccinic acid or N-acetyl cysteine. Free Radic Biol Med 21:157–161
Foster KA, Galeffi F, Gerich FJ, Turner DA, Müller M (2006) Optical and pharmacological tools to investigate the role of mitochondria during oxidative stress and neurodegeneration. Prog Neurobiol 79:136–171
Franco R, Sánchez-Olea R, Reyes-Reyes EM, Panayiotidis MI (2009) Environmental toxicity, oxidative stress and apoptosis: Ménage à trois. Mutat Res 674:3–22
Fujita T, Inoue H, Kitamura T, Sato N, Shimosawa T, Maruyama N (1998) Senescence marker protein-30 (SMP30) rescues cell death by enhancing plasma membrane Ca2+-pumping activity in HepG2 cells. Biochem Biophys Res Commun 250:374–380
Goyer RA (1989) Mechanisms of lead and cadmium nephrotoxicity. Toxicol Lett 46:153–162
Hirpara JL, Clément MV, Pervaiz S (2001) Intracellular acidification triggered by mitochondrial-derived hydrogen peroxide is an effector mechanism for drug-induced apoptosis in tumor cells. J Biol Chem 276:514–521
Kaufman DS, Goligorsky MS, Nord EP, Graber ML (1993) Perturbation of cell pH regulation by H2O2 in renal epithelial cells. Arch Biochem Biophys 302:245–254
Koh JY, Choi DW (1987) Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods 20:83–90
Kong IC, Bitton G, Koopman B, Jung KH (1995) Heavy metal toxicity testing in environmental samples. Rev Environ Contam Toxicol 142:119–147
Liu Z (2003) Lead poisoning combined with cadmium in sheep and horses in the vicinity of non-ferrous metal smelters. Sci Total Environ 309:117–126
Loghman-Adham M (1997) Renal effects of environmental and occupational lead exposure. Environ Health Perspect 105:928–938
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Matsuyama S, Llopis J, Deveraux QL, Tsien RY, Reed JC (2000) Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat Cell Biol 2:318–325
Morales AI, Vicente-Sánchez C, Sandoval JM, Egido J, Mayoral P, Arévalo MA et al (2006) Protective effect of quercetin on experimental chronic cadmium nephrotoxicity in rats is based on its antioxidant properties. Food Chem Toxicol 44:2092–2100
Muntner P, Menke A, Batuman V, Rabito FA, He J, Todd AC (2007) Association of tibia lead and blood lead with end-stage renal disease: a pilot study of African–Americans. Environ Res 104:396–401
Nolan CV, Shaikh ZA (1992) Lead nephrotoxicity and associated disorders: biochemical mechanisms. Toxicology 73:127–146
Nouwen EJ, Dauwe S, Van der Biest I, De Broe ME (1993) Stage- and segment-specific expression of cell-adhesion molecules N-CAM, A-CAM, and L-CAM in the kidney. Kidney Int 44:147–158
Nriagu JO, Pacyna JM (1988) Quantitative assessment of worldwide contamination of air, water and soils by trace metals. Nature 333:134–139
Oh SH, Lim SC (2006) A rapid and transient ROS generation by cadmium triggers apoptosis via caspase-dependent pathway in HepG2 cells and this is inhibited through N-acetylcysteine-mediated catalase upregulation. Toxicol Appl Pharmacol 212:212–223
Ott M, Gogvadze V, Orrenius S, Zhivotovsky B (2007) Mitochondria, oxidative stress and cell death. Apoptosis 12:913–922
Patra RC, Swarup D, Dwidedi SK (2001) Antioxidant effects of α tocopherol, ascorbic acid and l-methionine on lead-induced oxidative stress of the liver, kidney and brain in rats. Toxicology 162:81–88
Peñuelas J, Filella I (2002) Metal pollution in Spanish terrestrial ecosystems during the twentieth century. Chemosphere 46:501–505
Qin XJ, Li YN, Liang X, Wang P, Hai CX (2008) The dysfunction of ATPases due to impaired mitochondrial respiration in phosgene-induced pulmonary edema. Biochem Biophys Res Commun 367:150–155
Sen T, Sen N, Tripathi G, Chatterjee U, Chakrabarti S (2006) Lipid peroxidation associated cardiolipin loss and membrane depolarization in rat brain mitochondria. Neurochem Int 49:20–27
Thévenod F (2009) Cadmium and cellular signaling cascades: to be or not to be? Toxicol Appl Pharmacol 238:221–239
Üner N, Oruç E, Sevgiler Y (2005) Oxidative stress-related and ATPase effects of etoxazole in different tissues of Oreochromis niloticus. Environ Toxicol Pharmacol 20:99–106
Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C (1995) A novel assay for apoptosis, flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein-labeled Annexin V. J Immunol Methods 184:39–51
Walker LA, Simpson VR, Rockett L, Wienburg CL, Shore RF (2007) Heavy metal contamination in bats in Britain. Environ Pollut 148:483–490
Wang XQ, Xiao AY, Sheline C, Hyrc K, Yang A, Goldberg MP et al (2003) Apoptotic insults impair Na+, K+-ATPase activity as a mechanism of neuronal death mediated by concurrent ATP deficiency and oxidant stress. J Cell Sci 116:2099–2110
Wang L, Chen D, Cao J, Liu Z (2009a) Protective effect of N-acetylcysteine on experimental chronic cadmium nephrotoxicity in immature female rats. Hum Exp Toxicol 28(4):221–229
Wang L, Cao J, Chen D, Liu X, Lu H, Liu Z (2009b) Role of oxidative stress, apoptosis, and intracellular homeostasis in primary cultures of rat proximal tubular cells exposed to cadmium. Biol Trace Elem Res 127:53–68
Wang L, Chen D, Wang H, Liu Z (2009c) Effects of lead and/or cadmium on the expression of metallothionein in the kidney of rats. Biol Trace Elem Res 129(1–3):190–199
Wang L, Wang H, Hu M, Cao J, Chen D, Liu Z (2009d) Oxidative stress and apoptotic changes in primary cultures of rat proximal tubular cells exposed to lead. Arch Toxicol 83:417–427
Wang L, Li J, Li J, Liu Z (2010) Effects of lead and/or cadmium on the oxidative damage of rat kidney cortex mitochondria. Biol Trace Elem Res 137:69–78
Włostowski T, Krasowska A, Bonda E (2008) Joint effects of dietary cadmium and polychlorinated biphenyls on metallothionein induction, lipid peroxidation and histopathology in the kidneys and liver of bank voles. Ecotoxicol Environ Saf 69:403–410
Yang CF, Shen HM, Shen Y, Zhuang ZX, Ong CN (1997) Cadmium-induced oxidative cellular damage in human fetal lung fibroblasts (MRC-5) cells. Environ Health Perspect 105:712–716
Acknowledgements
This work was supported by the Shandong Provincial Natural Science Foundation of China (Grant No. ZR2010CQ014) and Science and Technology Innovation foundation for young scholars of Shandong Agricultural University in China (Grant No. 23690). Furthermore, Lin Wang and Heng Wang contributed equally to this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, L., Wang, H., Li, J. et al. Simultaneous Effects of Lead and Cadmium on Primary Cultures of Rat Proximal Tubular Cells: Interaction of Apoptosis and Oxidative Stress. Arch Environ Contam Toxicol 61, 500–511 (2011). https://doi.org/10.1007/s00244-011-9644-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00244-011-9644-4