
Abstract
Lead is a known nephrotoxic element. In this study, primary cultures of rat proximal tubular (rPT) cells were treated with different concentrations of lead acetate (0.25, 0.5 and 1 μM) to investigate its cytotoxic mechanism. A progressive loss in cell viability together with a significant increase in the number of apoptotic and necrotic cells and lactate dehydrogenase release were seen in the experiment. Simultaneously, elevation of reactive oxygen species levels and intracellular [Ca2+]i, depletion of mitochondrial membrane potential and intracellular glutathione were revealed during the lead exposure. In addition, apoptotic morphological changes induced by lead exposure in rPT cells were demonstrated by Hoechst 33258 staining. The apoptosis was markedly prevented by N-acetyl-l-cysteine, while the necrosis was not affected. Moreover, catalase and superoxide dismutase activities in the living cells rose significantly. In conclusion, exposure of rPT cells to low-concentration lead led to cell death, mediated by an apoptotic and a necrotic mechanism. The apoptotic death induced by oxidative stress was the chief mechanism. Meanwhile, a group of cells survived lead action, mediated by their ability to activate antioxidant defense systems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lead is a toxic, non-degradable heavy metal that was added to petrol in the early twentieth century to improve fuel efficiency. Lead in petrol has caused more environmental lead exposure than any other source. The impact of lead exposure has been very high in countries where lead is still used in petrol or has only recently been phased out, especially in developing countries. In addition, the exposed population may get exposed to lead due to food and water contamination, occupational exposure, and air pollution caused by industrial emission (Kadir et al. 2008). The persistence of lead in the animals and humans and the associated health risk is a topic of current concern. It has been found to produce wide range of toxic-biochemical effects, e.g., lead poisoning negatively impacts various body systems particularly, the haematopoietic, central, and renal system (Ercal et al. 1996).
Oxidative stress and apoptosis (programmed cell death) are two important phenomena in lead-induced toxicity. Oxidative stress induced by lead in the soft tissues has been postulated to be one of the important mechanisms of its toxic effects. Many animal studies have showed that lead is capable of causing oxidative stress in the kidney, liver, and brain (Ercal et al. 1996; Patra et al. 2001). Disruption of prooxidant/antioxidant balance induced by lead could lead to the tissue injury. Moreover, recent report also indicated that apoptosis might be associated with the lead-induced cytotoxicity (Pulido and Parrish 2003). Numerous studies demonstrate that lead exposure is capable of inducing apoptosis in various cell types (Fox et al. 1998; Cheng et al. 2002; Xu et al. 2006). Mitochondrial alterations play a central role in the apoptosis induced by lead (Fox et al. 1998). However, the mechanisms of lead-induced apoptosis are still largely unknown so far. Also, the correlation between oxidative stress and apoptosis in the toxic effects of lead has not yet been defined explicitly. Thus, combined effects of oxidative stress and apoptosis in the lead toxicity need further study.
The kidney is a sensitive target organ for lead exposure. The nephrotoxic effects of lead may occur at very low levels. Several epidemiologic studies found a higher prevalence of chronic kidney disease at blood lead levels of 10 μg/dl (0.48 μM) to 80 μg/dl (3.85 μM), while high blood lead levels (>80 μg/dl) are rare nowadays (Loghman-Adham 1997; Muntner et al. 2007; Kadir et al. 2008). The nephrotoxicity of lead has been extensively studied and widely reported in occupationally (Loghman-Adham 1997) and environmentally exposed human subjects (Muntner et al. 2007; Kadir et al. 2008), as well as in various experimental models (Ercal et al. 1996; Patra et al. 2001). Most studies on the nephrotoxicity of lead were implicated in oxidative stress and the role of lead-induced apoptosis in nephrotoxicity was little referred. Furthermore, most experiments related to lead nephrotoxicity concentrated on animal studies, in vitro studies were little reported. Cell cultures are valuable tools for mechanistic studies at the cellular level. Therefore, the intent of this paper was to set up an in vitro system to study lead nephrotoxicity. Due to the fact that primary cultures can better represent the live tissue than cultures of permanent cell lines, they are ideal for in vitro toxicity studies. Moreover, the renal tubule is one major site of renal impairment (Goyer 1989); a very pure preparation of isolated proximal tubular cells from rat kidneys was brought into culture. This cell model was set up to investigate the relationship between oxidative stress and apoptosis in the lead nephrotoxicity.
Materials and methods
Chemicals
All the chemicals were of highest grade purity available. Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Laboratories (Tokyo, Japan) and Annexin V-FITC Apoptosis Detection kit from Pharmingen (Becton Dickinson Company, USA). E-cadherin (L-CAM) was purchased from BD Transduction Laboratories (Lexington, KY). Fetal bovine serum (FBS) was purchased from Sijiqing Biological Engineering Material (Hangzhou, China). 5′-chloromethylfluorescein diacetate (CMF-DA), N,N,N′,N′-tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN) and Fluo-3/AM were purchased from Molecular Probes, USA. DMEM-F12 (1:1), lead acetate (PbAc2), propidium iodide (PI), 2′,7′-dichlorofluorescein diacetate (DCFH-DA), Hoechst 33258 staining, antibiotic–antimycotic solution, rhodamine 123 (Rh 123), collagenase, trypsin, N-acetyl-l-cysteine (NAC), EDTA/EGTA and all other chemicals were purchased from Sigma-Aldrich, USA.
Cell isolation and culture
Isolation of rat proximal tubular (rPT) cells has been described in detail elsewhere (Lühe et al. 2003), so the procedure will only be mentioned briefly. Male SD rats with a body weight between 180 and 200 g were anesthetized by an intraperitoneal injection of sodium pentobarbital [50 mg/kg body weight (bw)], and the kidneys were in situ perfused with Ca2+-free Hank’s balanced salt solution (HBSS) containing 0.5 mM EGTA. After removing the capsule, the kidneys were digested under permanently rotating conditions in a 200-U/ml collagenase IV-HBSS solution containing 4 mM CaCl2 and 1% penicillin/streptomycin for 15 min at 37°C. Kidneys were cut in half and the cortex was separated from the medulla by scraping it with a scalpel and cutting it into small pieces. Cells were isolated mechanically by squashing these pieces through gauzes of 135 μm followed by filtering through a 70-μm mesh. The isolated rPT cells were centrifuged at 700 rpm for 5 min at 4°C and washed once with the culture medium (DMEM/F12). Finally, cells were seeded into collagen I coated 6-well plates at a density of 5.0 × 105 cells per well. After an attachment phase of 72 h the medium was renewed the first time. Since then the medium was renewed every 2 days. The primary cells reached confluence after 144 h of cultivation and were subcultured after a 6-day culture by the trypsin–EDTA digestive method. Both primary cells and subcultures were cultured in DMEM/F12 medium supplemented with 25 mM HEPES, 10% FBS (heat-inactivated at 56°C), 0.25 g/L of glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin at 37°C in the presence of 95% air and 5% CO2. Cell viability was checked by the trypan blue exclusion and it was routinely higher than 95%. Identity of the proximal tubular cells has been confirmed by staining with antibodies against specific proximal tubular antigens. Staining with E-cadherin (L-CAM) antibody was carried out to confirm absence of other kidney cell types, such as distal tubular cells, collecting duct cells, and glomerulum-derived cells (Nouwen et al. 1993). The first passage was used to perform the experimental design when cultured for 120 h, which was in its highest cell viability (according to the growth curve, data not shown).
Lead treatment
In this study toxic effects of lead acetate, the most bioavailable form of inorganic lead salts, were investigated. Regarding the toxic concentrations chosen for this experiment, cells were treated with a range of lead acetate (PbAc2) concentrations (0, 0.25, 0.5, 1, 2, 4 μM) for 12 h, and cell viability was tested using MTT assay (data not shown). Several experiments from different cultures showed that IC50 value for rPT cells at 12 h was 2.18 μM and 4 μM Pb concentration leads to 100% cell death. Based on blood lead levels of lead-exposed patients reported in epidemiologic studies (Loghman-Adham 1997; Muntner et al. 2007; Kadir et al. 2008), low concentrations of lead acetate were used in this study. That is, cell cultures were incubated in the presence of 0, 0.25, 0.5, and 1 μM PbAc2, in a serum-free medium at different time intervals. Primarily, events of lead exposure over a 12-h period were chosen to investigate the lead nephrotoxicity.
Determination of cell survival
Cell Counting Kit-8 (CCK-8) is a one-bottle solution, which contains water-soluble tetrazolium salt. It can reduce the dehydrogenase in the mitochondria to water-soluble formazan dyes. The absorbance of these formazan dyes at 450 nm is proportional to the number of viable cells in the medium. Cells were seeded at a density of 1 × 104 in 96-well plates. After the preprocessing, the cells were treated with various Pb concentrations (0.25, 0.5 and 1 μM) for a time range of 3, 6, 12, and 24 h. Also, the cells were incubated with different concentrations of lead and 100 μM NAC for 12 h to assess the cytoprotective effect of NAC on cell survival. At the destined time points, cell viability assays were performed using CCK-8, according to manufacturer’s instructions. The absorbance was read at 450 nm by the microplate reader (Sunrise, Austria).
Lactate dehydrogenase (LDH) release
LDH activity was measured as the rate of decrease of the absorbance at 340 nm, resulting from the oxidation of NADH to NAD (Koh and Choi 1987). For the determination of LDH, the culture medium was collected after 12 h of lead treatment and the adherent cells on the plates were washed with phosphate-buffered saline (PBS) and then lysed with 0.1 M Tris–HCl (pH 7.4) containing 0.1% Triton X-100. LDH activity was measured both in culture medium and in cell lysate, following the addition of 1 mM pyruvate and 0.2 mM β-NADH. LDH release was given as percentage of LDH in the culture medium with respect to the total LDH (the sum between LDH in the culture medium and LDH inside the cells).
Hoechst 33258 staining
Apoptotic morphological changes in the nuclear chromatin were detected by staining with the DNA binding fluorochrome Hoechst 33258 (bisbenzimide). rPT cells were seeded on sterile cover glasses placed in the 24-well plates. After incubation with various concentrations of Pb (0.25, 0.5 and 1 μM) for 12 h, cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min, and then incubated with 50 μM Hoechst 33258 staining solution for 10 min. After three washes with PBS, the cells were viewed under an inverted fluorescence microscope (Olympus, IX-70, Japan) at an excitation wavelength of 352 nm through FITC filter (blue fluorescence). For assessing the extent of the apoptosis induced by lead treatment, 200 cells were randomly selected to count those apoptotic cells within every batch of experiment, each one performed in triplicate.
Flow cytometric analysis
All the following assays were carried out on BD-FACS Aria flow cytometer and the 488-nm laser was utilized. Cells were seeded at a density of 3 × 105 in 6-well plates and treated with various Pb concentrations (0.25, 0.5 and 1 μM) for 12 h. In addition, 100 μM NAC was co-administered with Pb for 12 h to assess its effect on apoptosis. After the treatment, the adherent cells were collected with trypsin–EDTA solution (1.25 g/L trypsin, 0.2 g/L EDTA). The detached and adherent cells were pooled and harvested by centrifugation at 1,500 rpm for 5 min. The harvested cells were washed twice with PBS and incubated with various fluorescent dyes for the flow cytometric analysis. Cell debris, characterized by a low FSC/SSC was excluded from analysis. The data were analyzed by Cell Quest software and mean fluorescence intensity was obtained by histogram statistics.
Assessment of apoptotic and necrotic cells
The apoptotic and necrotic cell distribution was analyzed by annexin V binding and propidium iodide (PI) uptake. Positioning of quadrants on annexin V/PI dot plots was analyzed by Cell Quest software and living cells (annexin V−/PI−), early apoptotic/primary apoptotic cells (annexin V+/PI−), late apoptotic/secondary apoptotic cells (annexin V+/PI+) and necrotic cells (annexin V−/PI+) were distinguished (Vermes et al. 1995). Therefore, the total apoptotic proportion included the percentage of cells with fluorescence annexin V+/PI− and annexin V+/PI+. After the exposure (12 h), 400 μl of binding buffer (1×) was added to the harvested cells of each sample. The resuspended cells were incubated with 10 μl annexin V-FITC and 20 μl PI for 15 min in dark at room temperature. The FITC and PI fluorescence was measured through FL-1 filter (530 nm) and FL-2 filter (585 nm), respectively, and 10,000 events were acquired.
Mitochondrial membrane potential (ΔΨ) determination
For the detection of mitochondrial ΔΨ, the harvested cells were incubated with Rh 123 (5 μg/ml final concentration) for 30 min in dark at 37°C, harvested and resuspended in PBS. The mitochondrial ΔΨ was measured by the fluorescence intensity (FL-1, 530 nm) of 10,000 cells.
Reactive oxygen species (ROS) measurement
Generation of ROS was monitored by measurement of hydrogen peroxide (H2O2) generation using the fluorescent probe DCFH-DA. This dye is cleaved to form non-fluorescent dichlorofluorescein (DCFH) in the cells, which gets oxidized to fluorescent dichlorofluorescein (DCF) by ROS. Thus, the DCF fluorescence intensity is proportional to the amount of peroxides produced by the cells. Briefly, 1.5 × 106 harvested cells/mL was incubated with DCFH-DA (100 μM final concentration) for 30 min in dark at 37°C. The incubated cells were harvested and suspended in PBS and ROS generation was measured by the fluorescence intensity (FL-1, 530 nm) of 10,000 cells on flow cytometer.
Analysis of intracellular free Ca2+ concentration ([Ca2+]i)
Following the treatment, the harvested cells were incubated with 0.5 mmol/L of TPEN (lead chelator) to discount Pb interference in the fluorescence signal, because calcium and lead interact similarly with the Fluo-3/AM dye (Dyatlov et al. 1998). Then, the cells were loaded with Fluo-3/AM (1 μM final concentration) for 30 min in dark at 37°C, and then washed with D-Hank’s balanced salt solution (Ca2+-free and Mg2+-free). Intracellular [Ca2+]i levels were represented with fluorescent intensity (FL-1, 530 nm) of 10,000 cells on flow cytometer.
Glutathione (GSH) measurement
The cellular level of GSH was monitored by CMF-DA. This fluorescent probe reacts with intracellular sulfhydryls and reflects predominantly GSH (Chikahisa et al. 1996). The harvested cells were incubated with CMF-DA (1 μM final concentration) for 30 min in dark at 37°C. The incubated cells were harvested, suspended in PBS and GSH was measured by the fluorescence intensity (FL-1, 530 nm) of 10,000 cells (Okada et al. 2000).
Antioxidant enzyme detection
Cells were seeded at a density of (5–6) × 105 in 4-well plates and treated with different concentrations of lead acetate (0.25, 0.5 and 1 μM) for 12 h. After the treatment, the adherent cells (live cells) were collected to detect the antioxidant enzymes.
Measurement of catalase (CAT) activity
After lead treatment, 50 mM of potassium phosphate (pH 7.2) was added to the collected cells, and then the samples were sonicated. The cellular suspension was centrifuged in an Eppendorf tube at 13,000 rpm for 4 min and at 4°C, and CAT activity was measured in the supernatant (López et al. 2006). Results were expressed as increment of optical density per minute per milligram protein.
Superoxide dismutase (SOD) activity
After lead treatment, 50 mM Tris–KCl (pH 8.2) was added to the collected cells, and then sonicated. The cellular suspension was centrifuged at 13,000 rpm for 4 min at 4°C. SOD activity was measured in supernatants according to Marklund and Marklund (1974).
Data presentation
Data are presented as mean ± SD from at least three independent experiments with different batches of cells, each one performed in duplicate or triplicate. Statistical comparisons were made using one-way analysis of variance (ANOVA) (Scheffe’s F test) after ascertaining the homogeneity of variance between the treatments. Differences were considered significant at P < 0.05.
Results
Action of lead on cellular viability
Lead caused a progressive cellular death in rPT cells, exhibited a concentration-dependent and time-dependent fashion during the exposure (Fig. 1a). Whereas 1 μM Pb effectively affected cell viability after a 3-h incubation period (P < 0.05), the lowest concentration tested, 0.25 μM Pb, significantly decreased the cell viability after a 12-h exposure time (P < 0.01). The cellular death caused by 1 μM Pb was always higher than that by 0.25 μM Pb or 0.5 μM Pb at the same exposure time. At the longest exposure time (24 h), the 0.25 μM Pb concentration caused ~9% loss in cell viability. That is, almost 91% cells remained viable as compared to ~72% at 1 μM Pb. In addition, the cellular death induced by lead (0.25, 0.5 and 1 μM) at 12 h was prevented, in part, by NAC (100 μM), an antioxidant agent (P < 0.05) (Fig. 1b). However, NAC treatment alone did not affect the cell viability (P > 0.05).

Effects of lead acetate on cell viability a and protection against cellular death by NAC b, measured by cck-8 reduction method. a The cells were incubated with increasing concentrations of PbAc2 for different periods of time. Cell viabilities of the control group at the destined time points were assumed as 100% and the variations were represented as percentage. Absorbance was measured at 450 nm. Each point represents mean ± SD (n = 6).* P < 0.05, ** P < 0.01 as compared to respective control, using one-way ANOVA. b The cells were incubated with PbAc2 and/or 100 μM NAC for 12 h to determine the cytoprotective action. Two different colors were chosen to point out which cells were treated with NAC (+) and which were not (−); i.e., white columns for no NAC-treatment and black for NAC-treatment. Data are mean ± SD (n = 6). * Statistical significance between control and lead treatment (0.25, 0.5 and 1 μM PbAc2) among the no NAC-treated groups; #Statistical significance between cells cultured in the absence (−) and presence (+) of NAC. ns Not significant; ** P < 0.01; # P < 0.05
Induction of necrosis and apoptosis by lead
The cellular death induced by lead was performed by the necrotic mechanism judging by the LDH release (Fig. 2a). After 12 h exposure, the necrosis induced by lead acetate (0.25, 0.5 and 1 μM) increased significantly (P < 0.01). However, the necrosis was not inhibited by the antioxidant molecule (NAC) (P > 0.05). The apoptosis induced by lead was performed by flow cytometric analysis. The annexin V binding assay measures the fluorescence generated by annexin binding with externalized phosphatidylserine of apoptotic cells. Also, the necrotic cells can be reflected when the annexin V binding assay incorporates PI. As shown in Fig. 2b, the number of apoptotic cells enhanced progressively with the lead concentration. At 12 h, the proportion of apoptotic cells drastically increased from 3.2% (control) to 48.9% at 1 μM Pb (P < 0.01), while the apoptosis was significantly prevented by NAC (P < 0.05). The number of necrotic cells was incremental with increase in lead concentration as shown in Fig. 2b. All the same, the necrosis was not inhibited by NAC (P > 0.05). Meanwhile, NAC treatment alone did not affect the necrosis and apoptosis (P > 0.05).These results indicate that lead acetate induced apoptosis and necrosis in rPT cells.

Lead action on cellular death type. Effect of NAC on necrosis a and b, measured by LDH release and flow cytometry; and apoptosis b, measured by flow cytometry. The cells were incubated with PbAc2 and/or 100 μM NAC for 12 h to measure the assays. Two different colors were chosen to point out which cells were treated with NAC (+) and which were not (−); i.e., white columns for no NAC-treatment and black for NAC-treatment. Results are mean ± SD of three separate experiments and each one performed in triplicate. *Statistical significance between control and lead treatment (0.25, 0.5 and 1 μM PbAc2) among the no NAC-treated groups; #Statistical significance between cells cultured in the absence (−) and presence (+) of NAC. ns Not significant; * P < 0.05, ** P < 0.01; # P < 0.05
Effect of lead on apoptotic morphological changes
Apoptotic morphological changes induced by lead were assessed by using Hoechst 33258 staining (Fig. 3a). In control group, the majority of cells had uniformly stained nuclei and the chromatin of normal nuclei was unaltered and spread uniformly throughout the entire nucleus. After exposure to lead acetate (0.25, 0.5 and 1 μM) for 12 h, it showed morphological changes typical of apoptosis, i.e., nuclear chromatin condensed and fragmented chromatin was characterized by a scattered, drop-like structure. The nuclei of apoptotic cells appeared smaller and shrunken when compared to intact cells. In addition, the statistical result (Fig. 3b) of cells with apoptotic characteristics shows that the number of apoptotic cells enhanced progressively with the lead concentration.

Effects of lead on apoptotic morphological changes in rPT cells. Cells were incubated with PbAc2 (0.25, 0.5 and 1 μM) for 12 h and nuclear chromatin changes (apoptosis) were assessed by Hoechst 33258 staining. a Among the groups, a control; b 0.25 μM Pb; c 0.5 μM Pb; d 1 μM Pb. In lead exposed groups, changes of nuclei fragmentation with condensed chromatin are evident (thin arrows). b The statistical results of apoptotic cells are expressed as mean ± SD of three separate experiments and each one performed in triplicate. ** P < 0.01 as compared to control, using one-way ANOVA
Effect of lead on mitochondrial membrane potential (ΔΨ)
Rh 123, a lipophilic cationic fluorescent dye, is selectively taken up by mitochondria and used to assess the mitochondrial ΔΨ of rPT cells. As shown in Fig. 4a, the mitochondrial ΔΨ was decreased substantially (P < 0.01) in rPT cells after exposure to lead acetate (0.25, 0.5 and 1 μM) for 12 h. Also, its reduction was in a concentration-dependent manner.

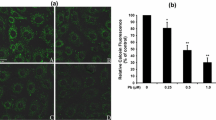
Effects of lead on mitochondrial membrane potential a, generation of ROS b, intracellular [Ca2+]i levels c and intracellular GSH levels d, measured by flow cytometry. a Cells were treated with PbAc2 (0.25, 0.5 and 1 μM) for 12 h. Rh 123 was added and the harvested cells were incubated for 30 min. The fluorescence was measured using flow cytometer with FL-1 filter. b The harvested cells were incubated with 100 μM DCFH-DA for 30 min at 37°C. DCF fluorescence was measured using flow cytometer with FL-1 filter. c The collected cells were first incubated with 0.5 mmol/L of TPEN, and then the harvested cells were incubated with 1 μM Fluo-3/AM for 30 min at 37°C. Fluo-3 fluorescence was measured using flow cytometer with FL-1 filter. d The harvested cells were incubated with 1 μM CMF-DA for 30 min in dark at 37°C. CMF fluorescence was measured using flow cytometer with FL-1 filter. Two different colors were chosen to point out which cells were treated with NAC (+) and which were not (−); i.e., white columns for no NAC-treatment and black for NAC-treatment. Fluorescence results were expressed as mean fluorescence intensity. Each bar represents mean ± SD (n = 4). *Statistical significance between control and lead treatment (0.25, 0.5 and 1 μM PbAc2) among the no NAC-treated groups; #Statistical significance between cells cultured in the absence (−) and presence (+) of NAC. ns Not significant; * P < 0.05, ** P < 0.01; # P < 0.05
Effect of lead on ROS generation and intracellular [Ca2+]i
The DCF fluorescence, proportionate to the ROS levels in the cells, was monitored on flow cytometer (Fig. 4b). The concentration-dependent generation of ROS increased at 12 h and the 0.25, 0.5 and 1 μM Pb concentration exhibited a 2.8-fold, 3.5-fold, and 4.3-fold significant (P < 0.01) increase, respectively, compared with the control group. Simultaneously, there was a concentration-dependent enhancement in intracellular [Ca2+]i (Fig. 4c). Significant [Ca2+]i increment in three Pb concentrations (0.25, 0.5 and 1 μM) was observed at 12 h (P < 0.05, P < 0.01).
Lead action on intracellular GSH levels and activities of CAT and SOD
After incubation with PbAc2 (0.25, 0.5 and 1 μM) and/or NAC (100 μM) for 12 h, significant GSH depletion caused by lead treatment is shown in Fig. 4d (P < 0.05), following by a concentration-dependent manner. However, the reduction of GSH levels after lead acetate exposure was markedly prevented by NAC (P < 0.05) (Fig. 4d). Information on activities of CAT and SOD in living cells is shown in Figs. 5a and b. At 12 h, it was found that lead significantly increased the CAT activity (P < 0.01). Simultaneously, there was a concentration-dependent significant increase in SOD activity that was caused by lead acetate (P < 0.01).

Effects of lead acetate on CAT activity (a) and SOD activity (b). rPT cells were treated for 12 h with the indicated lead concentrations. Data are mean ± SD of three experiments from cells of different cultures, each one performed in triplicate. ** P < 0.01 as compared to control, using one-way ANOVA
Discussion
Nephrotoxicity is one of the major side effects of exposure to xenobiotics, of which lead is a potent toxic agent that causes damage to the proximal tubular epithelium (Goyer 1989). Herein, this study was designed to investigate the intracellular events leading to PbAc2-induced nephrotoxicity in primary cultures of rat proximal tubular cells, focusing on the relationship between apoptosis and oxidative stress. Primarily, events of lead exposure over a 12-h period were chosen to investigate its toxic effects.
The progressive loss in cell viability shows that lead exposure induced cellular death in these cells, depending on both the lead concentration and the exposure time (Fig. 1a). The death was, in part, prevented by NAC (Fig. 1b), an antioxidant agent, which indicated that oxidative stress could be implicated in the mechanism by which lead acetate induces cell death in rPT cells. According to the results (Fig. 2), cellular death induced by lead is mediated by two mechanisms, necrotic and apoptotic. The increment in the LDH release rates (Fig. 2a) and increase in the number of necrotic cells (Fig. 2b) during the lead treatment were very remarkable, indicating cytosolic membrane breaking. It could be demonstrated that a necrotic mechanism played a role in the cellular death. On the other hand, significant increase in the number of apoptotic cells induced by lead was implicated with the apoptotic death (Fig. 2b). Compared with the apoptotic cells, the number of necrotic cells was small. In other words, the apoptotic death played a chief role in the cellular death induced by lead at these concentrations (0.25–1 μM). The apoptosis induced by lead was prevented by NAC significantly (Fig. 2b), while the necrosis was not affected by this antioxidant molecule (Figs. 2a, b). It also appears that in the apoptotic death mediated by lead, but not in the necrotic death, oxidative stress could be implicated. Thus, this study showed a certain type of relationship between apoptosis and oxidative stress in the toxic effects of lead on rPT cells. The necrotic death induced by lead was not discussed here, which will be worthy of further investigation.
In addition, lead-induced apoptosis was assessed by Hoechst 33258 staining (Fig. 3). Compared with the intact cells, lead exposure resulted in obvious apoptotic morphological changes (Fig. 3a). The percentage of apoptotic cells (Fig. 3b) was significantly increased in a concentration-dependent manner after lead treatment. This change tendency is consistent with the result obtained by flow cytometric analysis. Apoptotic morphological changes further show that apoptosis played a critical role in low concentration lead-induced cytotoxicity in rPT cells.
Moreover, the ability of lead to induce oxidative stress in rPT cells is aided by the induction of ROS by this cation. These cells treated with different concentrations of lead acetate (0.25, 0.5 and 1 μM) generated ROS significantly (Fig. 4b), demonstrating further that oxidative stress played a critical role in the lead toxicity. Mitochondria are the major source of ROS and its production is related to the level of electron transport (Fleury et al. 2002). ROS production rises when electron transport is reduced, which occurs in pathological situations (Wallace 2005). Large amounts of ROS seem to be lethal by inducing the mitochondrial permeability transition, and, as a result, mitochondria swelling, loss of the mitochondrial ΔΨ and further ROS production (Fleury et al. 2002; Foster et al. 2006). Then, the excessive ROS contribute to cell aging because they can oxidize lipids, proteins, and DNA (Chen et al. 2001). In this study, the induction of ROS in the rPT cells could be mediated by mitochondria alterations because lead produced a breakdown of the mitochondrial ΔΨ (Fig. 4a). ROS are not only inducers of cell death due to their high toxicity but these molecules also act as signaling molecules in apoptosis, which can directly activate apoptosis (Hervouet et al. 2007). In metal-induced apoptosis, it is thought that the mitochondria are most pertinent in mediating apoptosis, putatively via metal-induced ROS (Chen et al. 2001). Since accumulation of ROS targeted the mitochondrial membrane to induce a collapse of mitochondrial ΔΨ, it is reasonable to assume that the mitochondria play a crucial role in lead-induced apoptosis. This conclusion is consistent with previous results (Pulido and Parrish 2003).
Furthermore, evidences suggest that the elevation of intracellular [Ca2+]i is associated with the development of apoptosis (Orrenius et al. 1992; Rekasi et al. 2005). In this study, to evaluate the effect of lead on [Ca2+]i in rPT cells, Fluo-3/AM was chosen to use as an intracellular free Ca2+ fluorescent probe. We observed that treatment with PbAc2 (0.25, 0.5 and 1 μM) resulted in abnormal manifestation in intracellular [Ca2+]i, showing significant stronger [Ca2+]i existed in the treated cells than in control cells at 12 h (Fig. 4c). The fact suggests that abnormal Ca2+ homeostasis due to lead exposure may be another important mechanism of the development of apoptosis in these cells. Intracellular calcium overload may be related to the mitochondrial dysfunction. Since mitochondria are the major site of ATP production and mitochondrial ΔΨ is the driving force of ATP synthesis, a breakdown in the mitochondrial ΔΨ could lead to the fall in the ATP levels (Chakraborti et al. 1999). The resulting reduction in cellular ATP levels can lead to a disruption of ionic homeostasis which can cause an increase in [Ca2+]i and subsequent cellular apoptosis/necrosis (Grammatopoulos et al. 2004). Moreover, mitochondria act as temporary sinks to maintain the intracellular Ca2+ levels, which protect cells from toxic Ca2+ levels before releasing the Ca2+ into the cytosol (Nicholls 1985). Regarding the effect of mitochondria on the cell viability, the threatening event is the mitochondrial permeability transition, which occurs in response to calcium overload during elevated cellular ROS levels (Crompton 1999). The decrease of mitochondrial ΔΨ (Fig. 4a) and the increase of intracellular [Ca2+]i (Fig. 4c) induced by lead in rPT cells indicate a clear mitochondrial dysfunction together with an alteration of Ca2+ homeostasis. These results are in agreement with other investigations that indicate that nephrotoxicity is associated with many Ca2+-dependent processes (Alvarez-Barrientos et al. 2001), and that mitochondrial dysfunction seems to play an important role in several types of xenobiotics-induced nephrotoxicity (Aleo et al. 1991; Alvarez-Barrientos et al. 2001).
A growing body of evidence has indicated that intracellular redox status is one of the key mediators of apoptosis in many cell systems (Voehringer 1999; Schafer and Buettner 2001). A more highly oxidizing environment in the cell leads to apoptosis easily (Voehringer 1999). GSH is considered to be the major redox buffer of the cell (Schafer and Buettner 2001). It also has been demonstrated that GSH can protect cells against apoptosis—the protective role originates from multifactorial mechanisms that involve detoxification and modulation of cellular redox state and the subsequent redox-sensitive cell-signaling pathways and interaction with pro- and anti-apoptotic signals (Masella et al. 2005). In accord with these features, the increase of intracellular GSH might prevent apoptosis. In these cells treated with lead, NAC, an antioxidant molecule, was chosen to investigate the anti-apoptotic effect due to its ability to raise intracellular GSH levels (Lavrentiadou et al. 2001). The results indicated that lead-induced apoptosis was prevented by 100 μM NAC significantly (Fig. 2b). Moreover, lead treatment promoted a concentration-dependent decrease of GSH in rPT cells while the GSH depletion was significantly altered by NAC (Fig. 4d). This phenomenon is related to GSH synthesis as a protective mechanism. When the cells are oxidatively challenged, GSH synthesis increases as a protective mechanism. But with the higher concentration of toxicant, the GSH synthesis is unable to compete with oxidative stress, and so the levels tend to decline (Schafer and Buettner 2001). In a word, these results seem to suggest that oxidative stress induced by lead acetate in rPT cells is able to trigger apoptosis through the depletion of cellular GSH.
Beside cellular death mediated by lead ion, there are some surviving cells which are resistant to the action of this cation. The cellular survival in the presence of the toxic effect of ROS formation is based on the equilibrium between the toxic action and the ability of living cells to protect them against ROS action. Taking this into account, the possible antioxidative defense mechanisms in the living cells was investigated. SOD is one of the most effective intracellular enzymatic antioxidants, which catalyzes the dismutation of O −2 to O2 and to the less-reactive species H2O2. CAT is located in the peroxisomes, which very efficiently promotes the conversion of hydrogen peroxide to water and molecular oxygen (Matés et al. 1999). Our results indicate that in rPT cells, exposure to lead induced an increase in CAT (Fig. 5a) and SOD (Fig. 5b) activities at three lead concentrations. The results seem to suggest that there is a group of cells which are resistant to lead because they are able to induce enzymes against oxidative stress which produces the ROS. Maybe the increase in CAT and SOD activities is an indication of increased ROS generation in living cells due to lead treatment and this lend credence to the data in Fig. 4b. Also, the increases in these enzymatic activities could be due to an increase in the enzyme expression, since it is known that oxidative damages induce a cellular response, which tries to compensate the overload of the ROS formation (Dalton et al. 1999). Another possibility is that the enzyme activity was activated by the lead ion directly.
As far as the effect of lead on antioxidant enzymes is concerned, there are contradictory results (Sandhir et al. 1994; Sandhir and Gill 1995; Sivaprasad et al. 2003). Contrary to the present findings, lower activities of SOD and CAT were strongly correlated with lead intoxication in previous evidences. This diversity of results may be because the effect of lead varies in function according to (a) the cellular type, (b) the lead concentration used, and (c) the exposure time. Those variations might be due to the fact that the higher concentration of lead acetate and longer exposure time used in those studies compared to the lower concentration (0.25–1 μM) and the shorter exposure time (12 h) used in the present investigation. Also, most previous experiments were based on animal studies. Whether lead acetate influences the activities of antioxidant enzymes differently at low and high concentrations/for long and short exposure time is an area worthy of investigation in future studies.
In summary, oxidative stress plays a critical role in the cellular death induced by lead acetate in rPT cells. Apoptosis triggered by oxidative stress was the chief death mechanism in these cells. This death mechanism was produced by mitochondrial lead toxicity with breakdown of mitochondrial ΔΨ, elevation of intracellular [Ca2+]i, and ROS overproduction. Also, marked production of ROS and significant enhancement of intracellular [Ca2+]i indicate their crucial roles as intracellular mediators for the initiation of apoptotic events. In addition, depletion of cellular GSH further promoted the development of apoptosis. However, a group of cells survived. This may be due to the start of antioxidant mechanisms (activation of SOD and CAT) or other mechanisms, which will be worthy of further investigation.
References
Aleo MD, Wyatt RD, Schnellmann RG (1991) Mitochondrial dysfunction is an early event in ochratoxin a but not osoporein toxicity to rat renal proximal tubules. Toxicol Appl Pharmacol 107:73–80
Alvarez-Barrientos A, O’Connor JE, Nieto Castillo R, Moreno Moreno AB, Prieto P (2001) Use of flow cytometry and confocal microscopy techniques to investigate early CdCl2-induced nephrotoxicity in vitro. Toxicol Vitro 15:407–412
Chakraborti T, Das S, Mondal M, Roychoudhury S, Chakraborti S (1999) Oxidant, mitochondria and calcium: an overview. Cell Signal 11:77–85
Chen F, Vallyathan V, Castranova V, Shi X (2001) Cell apoptosis induced by carcinogenic metals. Mol Cell Biochem 222:183–188
Cheng YJ, Yang BC, Hsieh WC, Huang BM, Liu MY (2002) Enhancement of TNF-α expression does not trigger apoptosis upon exposure of glial cells to lead and lipopolysaccharide. Toxicology 78:183–191
Chikahisa L, Oyama Y, Okazaki E, Noda K (1996) Fluorescent estimation of H2O2-induced changes in cell viability and cellular nonprotein thiol level of dissociated rat thymocytes. Jpn J Pharmacol 71:299–305
Crompton M (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem J 341:233–249
Dalton TP, Shertzer HG, Puga A (1999) Regulation of gene expression by reactive oxygen. Annu Rev Pharmacol Toxicol 39:67–101
Dyatlov VA, Dyatlova OM, Parsons PJ, Lawrence DA, Carpenter DO (1998) Lipopolysaccharide and interleukin-6 enhance lead entry into cerebellar neurons: application of a new and sensitive flow cytometric technique to measure intracellular lead and calcium concentrations. Neurotoxicology 19:293–302
Ercal N, Treeratphan P, Hammond TC, Mattews RH, Grannemann N, Spitz D (1996) In vivo indices of oxidative stress in lead exposed C57BL/6 mice are reduced by treatment with meso-2, 3-dimercaptosuccinic acid or N-acetyl cysteine. Free Rad Biol Med 21:157–161
Fleury C, Mignotte B, Vayssière JL (2002) Mitochondrial reactive oxygen species in cell death signaling. Biochimie 84:131–141
Foster KA, Galeffi F, Gerich FJ, Turner DA, Müller M (2006) Optical and pharmacological tools to investigate the role of mitochondria during oxidative stress and neurodegeneration. Prog Neurobiol 79:136–171
Fox DA, He L, Poblenz AT, Medrano CJ, Blocker YS, Srivastava D (1998) Lead-induced alterations in retinal cGMP phosphodiesterase trigger calcium overload, mitochondrial dysfunction and rod photoreceptor apoptosis. Toxicol Lett 102–103:359–361
Goyer RA (1989) Mechanisms of lead and cadmium nephrotoxicity. Toxicol lett 46:153–162
Grammatopoulos TN, Johnson V, Moore SA, Andres R, Weyhenmeyer JA (2004) Angiotensin type 2 receptor neuroprotection against chemical hypoxia is dependent on the delayed rectifier K+ channel, Na+/Ca2+ exchanger and Na+/K+ ATPase in primary cortical cultures. Neurosci Res 50:299–306
Hervouet E, Simonnet H, Godinot C (2007) Mitochondria and reactive oxygen species in renal cancer. Biochimie 89:1080–1088
Kadir MM, Janjua NZ, Kristensen S, Fatmi Z, Sathiakumar N (2008) Status of children’s blood lead levels in Pakistan: implications for research and policy. Public health 122:708–715
Koh JY, Choi DW (1987) Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods 20:83–90
Lavrentiadou SN, Chan C, Kawcak T, Ravid T, Tsaba A, Der Vliet AV, Rasooly R, Goldkorn T (2001) Ceramide-mediated apoptosis in lung epithelial cells is regulated by glutathione. Am J Respir Cell Mol Biol 25:676–684
Loghman-Adham M (1997) Renal effects of environmental and occupational lead exposure. Environ Health Perspect 105:928–939
López E, Arce C, Oset-Gasque MJ, Cañadas S, González MP (2006) Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radic Biol Med 40:940–951
Lühe A, Hildebrand H, Bach U, Dingermann T, Ahr HJ (2003) A new approach to studying ochratoxin A (OTA)-induced nephrotoxicity: expression profiling in vivo and in vitro employing cDNA microarrays. Toxicol Sci 73:315–328
Marklund S, Marklund G (1974) Involvement of superoxide anion radical in the autooxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 47:469–474
Masella R, Benedetto R, Varì R, Filesi C, Giovannini C (2005) Novel mechanisms of natural antioxidant compounds in biological systems: involvement of glutathione and glutathione-related enzymes. J Nutr Biochem 16:577–586
Matés JM, Pérez-Gómez C, Núñez de Castro I (1999) Antioxidant enzymes and human diseases. Clin Biochem 32:595–603
Moran LK, Guteridge JM, Quinlan GJ (2001) Thiols in cellular redox signaling and control. Curr Med Chem 8:763–772
Muntner P, Menke A, Batuman V, Rabito FA, He J, Todd AC (2007) Association of tibia lead and blood lead with end-stage renal disease: a pilot study of African-Americans. Environ Res 104:396–401
Nicholls DG (1985) A role for the mitochondrion in the protection of cells against calcium overload? Prog Brain Res 63:97–106
Nouwen EJ, Dauwe S, Van der Biest I, De Broe ME (1993) Stage- and segment-specific expression of cell-adhesion molecules N-CAM, A-CAM, and L-CAM in the kidney. Kidney Int 44:147–158
Okada Y, Oyama Y, Chikahisa L, Satoh M, Kanemaru K, Sakai H, Noda K (2000) Tri-n-butyltin-induced change in cellular level of glutathione in rat thymocytes: a flow cytometric study. Toxicol Lett 117:123–128
Orrenius S, Mccabe MJ, Nicotera P (1992) Ca2+-dependent mechanisms of cytotoxicity and programmed cell death. Toxicol Lett 64:357–364
Patra RC, Swarup D, Dwidedi SK (2001) Antioxidant effects of α tocopherol, ascorbic acid and l-methionine on lead-induced oxidative stress of the liver, kidney and brain in rats. Toxicology 162:81–88
Pulido MD, Parrish AR (2003) Metal-induced apoptosis: mechanisms. Mutat Res 533:227–241
Rekasi Z, Czompoly T, Schally AV, Boldizsar F, Varga JL, Zarandi M, Berki T, Horvath RA, Nemeth P (2005) Antagonist of growth hormone releasing hormone induces apoptosis in LNCaP human prostate cancer cells through a Ca2+-dependent pathway. Proc Natl Acad Sci 102:3435–3440
Sandhir R, Gill KD (1995) Effect of lead on lipid peroxidation in livers of rats. Biol Trace Elem Res 48:91–97
Sandhir R, Julka D, Gill KD (1994) Lipoperoxidative damage on lead exposure in rat brain and its implications on membrane bound enzymes. Pharmacol Toxicol 74:66–71
Schafer FQ, Buettner GR (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Rad Biol Med 30:1191–1212
Sivaprasad R, Nagaraj M, Varalakshmi P (2003) Combined efficacies of lipoic acid and meso-2, 3-dimercaptosuccinic acid on lead-induced erythrocyte membrane lipid peroxidation and antioxidant status. Hum Exp Toxicol 22:183–192
Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C (1995) A novel assay for apoptosis, flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein-labeled Annexin V. J Immunol Meth 184:39–51
Voehringer DW (1999) Bcl-2 and glutathione: alterations in cellular redox state that regulate apoptosis sensitivity. Free Rad Biol Med 27:945–950
Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39:359–407
Xu J, Ji LD, Xu LH (2006) Lead-induced apoptosis in PC 12 cells: involvement of p53, bcl-2 family and caspase-3. Toxicol Lett 166:160–167
Acknowledgments
This work was supported by the National Nature Science Foundation of China (No. 30440050 and 30571364).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, L., Wang, H., Hu, M. et al. Oxidative stress and apoptotic changes in primary cultures of rat proximal tubular cells exposed to lead. Arch Toxicol 83, 417–427 (2009). https://doi.org/10.1007/s00204-009-0425-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-009-0425-z