
Abstract
Human endogenous retroviruses (HERVs) are the remnants of ancient germ cell infection by exogenous retroviruses and occupy up to 8% of the human genome. It has been suggested that HERV sequences have contributed to primate evolution by regulating the expression of cellular genes and mediating chromosome rearrangements. After integration ∼28 million years ago, members of the HERV-K (HML-2) family have continued to amplify and recombine. To investigate the utility of HML-2 polymorphisms as markers for the study of more recent human evolution, we compiled a list of the structure and integration sites of sequences that are unique to humans and screened each insertion for polymorphism within the human genome databases. Of the total of 74 HML-2 sequences, 18 corresponded to complete or near-complete proviruses, 49 were solitary long terminal repeats (LTRs), 6 were incomplete LTRs, and 1 was a SVA retrotransposon. A number of different allelic configurations were identified including the alternation of a provirus and solitary LTR. We developed polymerase chain reaction-based assays for seven HML-2 loci and screened 109 human DNA samples from Africa, Europe, Asia, and Southeast Asia. Our results indicate that the diversity of HML-2 elements is higher in African than non-African populations, with population differentiation values ranging from 0.6 to 9.8%. These findings denote a recent expansion from Africa. We compare the phylogenetic relationships of HML-2 sequences that are unique to humans and consider whether these elements have played a role in the remodeling of the hominid genome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Endogenous retroviruses (ERVs) are vertically transmitted genetic elements that remain from ancient retroviral infection of germ line cells. Following the original insertion of the provirus, intracellular retrotransposition and recombination have led to an increase in the copy number of particular families (Lower et al. 1996). ERVs are stably integrated into the genomes of all vertebrates and are transmitted as Mendelian genes. Analysis of the draft sequence of the human genome shows that approximately 8% is composed of retrovirus-like elements, which includes both proviral sequences and a large number of long terminal repeats (LTRs) (Lower et al. 1996; Patience et al. 1997; International Human Genome Sequencing Consortium 2001). Several distinct human ERV families (HERVs) have been identified, which show different genomic integration patterns (Urnovitz and Murphy 1996) and range in copy number from 1 to 1000 (Tristem 2000). HERVs are classified into families based upon their putative tRNA primer binding site specificity; HERV-I for Ile tRNA and HERV-K for lys tRNA. Mutational events have rendered most of these HERVs replication defective following integration, although many remain transcriptionally active (Goodchild et al. 1995; Huh et al. 2003). The HERV-K superfamily is acknowledged to be the most biologically active class of HERV, having retained the ability to encode functional retroviral protein (Towler et al. 1998) and produce retrovirus-like particles (Simpson et al. 1996; Seifarth et al. 1998).
Since the identification of the HERV-K prototype, HERV-K10 (Ono et al. 1986), phylogenetic analysis of a conserved reverse transcriptase (RT) region has led to the definition of six HERV-K subgroups, HML-1 to HML-6 (Medstrand and Blomberg 1993; Zsiros et al. 1998). The HML-2 group appears to have integrated into the germ line approximately 28 million years ago, before the evolutionary split of lower Old World primates and hominoids (Reus et al. 2001b). Despite this relative age, HML-2 open reading frames appear to be maintained (Zsiros et al. 1999) and the presence of sequences that are unique to humans indicates that they were continuing to undergo amplification relatively recently (Medstrand and Mager 1998; Barbulescu et al. 1999; Buzdin et al. 2002, 2003). HML-2 proviral genomes are classified into two types based upon a 292-bp deletion at the pol–env boundary, with Type I elements carrying the deletion. Both Type I and Type II proviral genomes have remained retrotranspositionally active following the evolutionary split of chimpanzees and hominids (Costas 2001). HML-2 elements are easily distinguished from their progenitor, HERV-K(OLD), as they have a 96-bp deletion in gag which has not disrupted the open reading frame and further 8- and 23-bp deletions within their LTRs (Mayer et al. 1998; Reus et al. 2001b).
Recent genome-wide comparisons of human and chimpanzee have demonstrated that large-scale genomic rearrangements, such as segmental duplications and the insertion of retroelements, provide a significant source of DNA variation within the host species (Liu et al. 2003; Frazer et al. 2003; Locke et al. 2003). To date, most evolutionary studies have focused on the interspersed repetitive elements, L1 (long interspersed element 1) and Alu (short interspersed element); these have shown that these retroelements serve as mutagens at both the structural and genic levels (Deininger and Batzer 2002). For the same reasons, HML-2 elements may also have contributed either by serving as nucleation points for homologous recombination (Hughes and Coffin 2001) or by regulating the expression of cellular genes (Lower et al. 1996; Akopov et al. 1998; Domansky et al. 2000; Vingradova et al. 2001). In this study we have examined the genomic structure and integration sites of HML-2 elements that are unique to humans and have investigated their potential role in the remodeling of the human genome. We have also analyzed their phylogeny and demonstrated their utility for the study of human genomic diversity.
Materials and Methods
Identification of HERV-K(HML-2) Polymorphisms
The GenBank nonredundant and high-throughput genomic sequence database (http://www.ncbi.nlm.nih.gov/genome/seq/HsBlast.html), the Ensembl database (http://www.ensembl.org), and the HERV-d (http://herv.img.cas.cz) database were screened using the HERV-K10 sequence (accession No. M14123) as a probe. Accessions containing full-length HML-2 genomes were aligned by hand in the SIMMONIC sequence analysis package (Simmonds and Smith 1999), with individual elements determined by their cellular flanking sequences and chromosomal location. The flanking regions of each genome were then screened by standard nucleotide–nucleotide BLAST against the nonredundant and high-throughput sequence databases, in order to detect paralogous sequences and to ascertain if polymorphism was present at specific loci. Accessions reported to contain human-specific HML-2 LTRs and near-complete genomes were also individually screened for polymorphism within the human genome databases, with subsequent designation according to their cytogenetic location and flanking sequences.
DNA Samples and PCR Primers and Conditions
Samples from a chimpanzee (Pan troglodytes) and gorilla (Gorilla gorilla) along with 25 African, 28 Asian, 22 European, and 34 Papua New Guinean humans were collected as buccal swabs or serum. Genomic DNA was isolated using the QIAamp DNA kit (Qiagen, UK), following the manufacturer’s instructions. DNA quality and authenticity were confirmed by PCR amplification for the sex chromosome-specific amelogenin gene (Faerman et al. 1995) on the human samples and the protamine gene on the chimpanzee samples (data not shown).
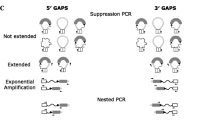
Each sample was subjected to a series of PCR amplification reactions in order to assess polymorphism within selected HML-2 loci (Fig. 1). DNA sequences adjacent to each HML-2 insertion were used to design unique flanking region primers. Primers were screened by standard nucleotide–nucleotide BLAST against the nonredundant and high-throughput sequence databases, to ensure that the DNA sequences were unique. Elements that resided in repetitive sequence regions could not be examined by PCR. Universal primers for HML-2 LTR, gag, and env genes were designed according to a consensus sequence, which was obtained by aligning all of the HML-2 sequences examined in this study. Heminested PCR reactions were performed in instances where single-round PCR proved difficult to optimize. This process utilized two consecutive rounds of amplification, the first round using an external pair of primers while the second round contained one of the first primers and a single nested primer which is internal to the first primer pair. The amplicon produced by the first round of PCR was used as a template for the second PCR amplification.

PCR scheme for detecting HERV-K(HML-2) allelic variants.
PCR amplification primers and conditions for each HML-2 loci are listed in Table 1. Reactions were carried out in volumes of 50 μl, with each containing 200 ng of genomic DNA, a 200 μM concentration of each dNTP, a 0.5 μM concentration of each primer, and 0.5 units of Taq DNA polymerase in standard PCR buffer as supplied by Promega. The second round of PCR used 2 μl of first-round PCR product and was performed in volumes of 30 μl, with a reaction mix as listed above. The resulting PCR products were analyzed by electrophoresis through a 2% agarose gel, with the product size confirmed by comparison to a 100-bp ladder (Promega). Nucleotide sequencing was carried out directly on second-round PCR products using ABI PRISM Big Dye kits (Applied Biosystems).
Sequence and Population Genetic Analysis
Sequence data obtained using the ABI PRISM kits were viewed using the CHROMAS sequence viewer and directly imported into the SIMMONICS sequence analysis package. Eighty-six full-length LTRs which were representative of 67 human-specific HML-2 insertions were aligned by hand in SIMMONIC. A neighbor-joining tree was constructed using MEGA, version 2.1 (http://www.megasoftware.net/), with the Kimura two-parameter distance estimate and pairwise deletion of gaps. Allele frequencies, Hardy–Weinberg tests, and Wright’s Fst statistic were estimated using PopGene, version 1.31 (http://www.ualberta.ca/∼fyeh/).
Results
Activity of HERV–K(HML-2) Elements Within the Human Lineage
Screening of the human sequence databases for HML-2 proviruses revealed 3 novel sequences and 29 formerly identified complete and near-complete elements (Table 2). A further 15 less intact proviral sequences have also previously been identified (Hughes and Coffin 2001; Reus et al. 2001b), bringing the total number of identified HML-2 proviruses within the human genome to 47. The three HML-2 near-complete proviruses identified in this study were located at 4p16 (AC105916), Xq28 (AF277315), and 10q24.2 (AL392107). The chimpanzee orthologue of the provirus contained within the pseudoautosomal region of the human X chromosome (Xq28) was also detected within accession AC144385.
Eight of 18 human-specific HML-2 proviral genomes were Type I and 10 were Type II, indicating the coexistence of two retrotranspositionally active master elements during hominid speciation. Further computational screening with the flanking regions of individual elements revealed that five were polymorphic, showing a number of different configurations (Fig. 1). Two proviruses, HERV-K113 and HERV-K115, were dimorphic for insertion, with one allele representing the presence and the second the absence of a complete provirus. Other variable features included the alternation of a complete provirus with a solitary LTR (HERV-K103 and HERV-K106) and, finally, the variable existence of a tandem duplication of the HERV-K108 provirus.
Human-specific HML-2 LTR sequences have previously been identified by targeted genomic difference analysis (TGDA) and BLAST determination, with subsequent phylogenetic identification by PCR amplification (Buzdin et al. 2002; Lebedev et al. 2000). We catalogued all of the human-specific HML-2 LTR sequences discovered to date, determined their cytogenetic location, and assigned subtype according to their classification in previous publications (Table 3). During sequence alignment we observed that several LTR sequences either had been observed by more than one study and were assigned different names or were misinterpreted as solitary LTRs when they were part of a complete provirus. Of the total of 74 human-specific HML-2 LTR sequences, 18 were complete or near-complete proviruses, 49 were solitary LTRs, and a further 6 could not be distinguished between near-complete proviral sequences and solitary LTRs, as they have lost the 5′ or 3′ end of their sequence. Further sequence comparison of the HML-2 LTR contained at Xq26.3 (AL359703) to the SVASTPA1 retroelement (AC016142) (Ostertag et al. 2003) revealed that this human-specific sequence was a member of the SVA (SINE, VNTR, and Alu) retrotransposon family. As SVA elements are derived from SINE.R retroelements which are composed of a partial HERV-K(HML-2) env and a 3′-LTR (Shen et al. 1994), it can be concluded that the LTR at Xq26.3 is not a direct product of the retrotransposition of a HERV-K (HML-2) provirus. Computational screening of the flanking regions of each of the human-specific HML-2 LTRs indicated that two solitary LTRs were polymorphic for insertion. The first was located at 6p21.32 (Z80898) and is reported to have arisen through duplication of the MHC complex (Horton et al. 1998); the second was located at 9q12 (AL39220). With the exception of chromosomes 13, 15, 18, and Y, all chromosomes contained at least one human-specific HERV-K(HML-2) LTR sequence that arose through the process of retrotransposition.
Inter- and Intraelement Recombination—Comparison of Direct Repeat Sequences
In common with infection by exogenous retroviruses, retrotransposition and subsequent integration of HERV-K endogenous retrovirus result in the generation of short target site duplications of 4 to 6 bp at the integration site. These direct repeat sequences flank either end of the newly integrated provirus and should be identical at the time of integration. Integrated proviruses or solitary LTRs with different target site duplications serve as potential signatures of interelement homologous recombination, which would be expected to have resulted in large-scale chromosomal rearrangements (Hughes and Coffin 2001). We examined the target site duplications of the 74 human-specific HML-2 sequences in order to investigate the impact of such events during hominid evolution. The three near-complete proviruses, 12q24.11 (AC002350); HERV-K(C19) (AF017229), and 21q21.1 (AL109763), along with the six incomplete LTRs, were not included in the data set, as they had lost one or more of their direct repeats. The human-specific HML-2 LTR at Xq26.3 (AL359703), which was a component of a SVA retroelement, and the polymorphic solitary LTR at 6p21.32 (Z80898) were also not included, as they did not represent the recent retrotranspostion and integration of a proviral sequence. This left a total of 63 HML-2 elements that could be analyzed.
Each of the respective allelic variants of the polymorphic loci, HERV-K103 and HERV-K106, had identical direct repeats, implying that the solitary LTRs were generated as a result of (intraelement) homologous recombination between the 5′- and the 3′-LTRs of each respective provirus. Such an event is also expected to produce and result in the loss of a near-complete provirus consisting of a single LTR along with the gag, pol, and env genes. For the same reasons, intraelement recombination leading to the internal duplication of a proviral sequence is also likely to have generated the tandem duplication of the HERV-K108 provirus and is expected to have also led to the formation of a solitary LTR at the same chromosomal location.
If HERV-K sequences with inconsistent direct repeats arose through (interelement) homologous recombination between proviruses located either on different chromosomes or in different regions of the same chromosome, then exchanges would be expected to produce a reciprocal HERV-K element with an opposite configuration of direct repeats and flanking regions. Of the remaining HML-2 sequences, two had disparate target site duplications, indicating their likely hybrid nature. Within the human genome databases they exist as solitary LTRs and are located on chromosomes 7p21.2 (AC006035) and 17q22 (AC032016).
We screened the human genome databases for the expected reciprocal product of each of the “hybrid” HML-2 sequences; none of the predicted sequences were present. This implied either that the reciprocal products were not present in the representative individuals sequenced by the human genome projects or that the expected reciprocal sequences do not form a constituent of the contemporary human gene pool. In order to confirm that the two human-specific solitary LTRs were a product of interelement recombination, we designed unique 5′ and 3′ flanking region primers for the solitary LTRs at 7p21.2 (AC006035) and 17q22 (AC032016) and conducted amplification for both the solitary LTR and the preintegration site in human and nonhuman primates. Initial results confirmed that both of the solitary LTRs were not present in chimpanzee and gorilla, indicating either that they were not fixed in the gene pool at the time of human/chimpanzee/gorilla divergence or that they had integrated during hominid evolution. We then performed amplification for the preintegration site in the nonhuman primates under the expectation that a negative result indicated interelement recombination. If recombination had occurred between different proviruses located at different chromosomal regions, then the expected product either would be too large to amplify or would not exist in nonhuman primates. Contrary to expectation, amplicons were produced, suggesting that the disparate target site duplications were generated by an alternative mechanism (Figs. 2 and 3).

Flanking region duplication leading to variable direct repeat sequences. A Solitary LTR 7p21.2. The preintegration sequence in chimpanzee and gorilla is represented by the top figure and contains a 250-bp sequence with the respective nucleotide sequences GGATGA and AGGCAA at each end. The human-specific solitary LTR at 7p21.2 (accession No. AC006035) has inconsistent direct repeat sequences AGGCAA and GGATGA, which are underlined. Sequence comparison indicates that the solitary LTR is flanked by a 250-bp duplication of the preintegration sequence. B Provirus 21q21.1. The top figure represents the preintegration sequence present in chimpanzee (accession No. BS000043). The near-complete human-specific provirus at 21p21.1 (accession No. AL109763) contains a truncated 3′-LTR of 257 bp which is adjacent to a 450-bp duplication of the preintegration site sequence.

Sequence alignment of the human-specific HERV-K (HML-2) solitary LTR at chromosomal location 17q22 and preintegration site in primates. The human sequence of the HERV-K(HML-2) LTR-containing locus (accession No. AC032016) is shown on the top line. Nucleotide substutions at each position are indicated with the appropriate nucleotide. Alignment gaps are indicated by dashes. The direct repeats of the solitary LTR are underlined.
Sequence analysis of the preintegration site and solitary LTR at 7p21.2 indicated that the variable direct repeats were a result of an apparent duplication of the 5′ flanking sequence (Fig. 2A). A similar situation was also observed for the human-specific provirus at 21q21.1, where the 3′-LTR of the provirus appears to be truncated by a sequence paralogous to the 5′ flanking sequence (Fig. 2B). Presuming that the provirus contained two identical LTRs at insertion, this duplication must have occurred following integration. For the solitary LTR at 17q22, sequence data on the preintegration site indicated that the downstream direct repeat was 4 bp shorter than the upstream one (Fig. 3). These observations indicate that inconsistent direct repeat sequences do not always reflect interelement recombination events (see Discussion).
Phylogeny of HML-2 LTRs Unique to Humans
In order to further examine the retrotransposition and evolution of the human-specific HML-2 elements, we generated a neighbor-joining tree from the alignment of 87 full-length HML-2 LTR sequences (Fig. 4). Individual LTR sequences are identified according to their consensus name or genomic location. With the exception of the HERV-K115 provirus, the 5′- and 3′-LTR of each individual HML-2 provirus grouped together, supporting the view that they had not undergone interelement recombination or sequence exchange. However, the LTRs of the HERV-K115 provirus were divergent, reflective of the provirus having undergone gene conversion. The solitary LTRs of the polymorphic loci HERV-K103 and HERV-K106 grouped with the LTRs of their progenitor provirus, confirming that they were generated through intraelement homologous recombination. The respective 5′- and 3′-LTRs of the tandemly repeated provirus and the single, provirus located at 7p22.1 (HERV-K108) also grouped, indicating that the tandem proviral sequence also arose through intraelement recombination. Interestingly, the distribution of the Type I and Type II proviral LTRs was not monophyletic, as would be expected if these elements follow a “master” or “source” model of retrotransposition (see Discussion).

Phylogeny of human-specific HML-2 LTRs. Individual LTRs are named according to the chromosomal location of the corresponding accession clone or bibliographic name of the sequence. P5—5-LTR; P3—3-LTR; S—Solitary LTR; R—direct repeats vary. The shaded LTRs are polymorphic and have arisen through intraelement recombination. The arrows emphasize the divergent LTRs of the HERV-K115 provirus. Roman numerals denote the genome structure of the HERV-K(HML-2) proviral sequences, with Type I sequences carrying a 292-bp deletion at the pol–env boundary. (○) LTR subfamily HS-a. (•) LTR subfamily HS-b.
HERV-K LTRs have previously been classified according to diagnostic nucleotide differences and intragroup divergence. Recently, 40 human-specific HML-2 LTR sequences were classified into the subtypes Hs-a and Hs-b (Buzdin et al. 2003). Superimposition of these subtypes on the phylogenetic tree indicated that this taxomony was consistent, although the two subtypes were not clearly distinguished and grouped independently of the Type I and Type II proviral genomes. This suggests that HML-2 LTRs may be subject to a high degree of sequence exchange between closely related sequences.
Relative Age of HML-2 Loci
During the retrotransposition of a provirus, reverse transcription generates a new retrovirus-like sequence containing two identical LTR sequences. Assuming that a provirus has not undergone any form of sequence exchange and there is no selective pressure acting on it, the accumulative nucleotide differences between the LTRs can serve as a molecular clock (Dangel et al. 1995). We calculated the number of nucleotide differences between the 5′- and the 3′-LTRs of the 32 intact HML-2 proviruses that were present within the human genome and compared each result to the relative age of the provirus (Table 2). Several inconsistencies were observed which were indicative of sequence homogenization between the LTRs of individual elements.
First, sequence data for individual elements were discrepant; this is exemplified by the human-specific provirus at 12q14.1 where the accession clone AC025420 has 4 nucleotide differences but the analogous accessions AC074261 and FID58908 had 19 and 20 differences, respectively. Second, relative age did not correspond with accumulative nucleotide differences. As the provirus at 11q23.2 (AP000831) is present in humans, chimpanzees, and gorillas, its relative age is expected to be between 6.2 and 12 mya (Chen and Li 2001). However, it contains only six accumulative nucleotide differences between the LTRs, which, compared to the relative age of comparable proviral loci such as 10p14 (AC015686), is indicative of a more recent insertion. Additionally, the LTRs of the insertionally polymorphic HERV-K113 provirus (AY037928) vary by 3 bp, whereas the HERV-K106 provirus (AC078785), which is reported to be universal in contemporary humans (Barbulescu et al. 1999), has only a 1-bp difference between the LTRs. In order to further investigate these discrepancies we designed PCR-based assays and tested for the absence of two human-specific proviral loci; 3q27.2 (AC069420) and HERV-K107 (5q33.3), which, according to their accumulative differences (three and two), appear to have integrated into the human germ line relatively recently.
PCR Analysis of HML-2 Loci
HERV-K polymorphisms serve as ideal genetic markers for examining human evolution, as they are stable and identical by descent and the ancestral state is known to be the absence of the insertion. We developed a PCR-based assay to examine the allelic variation associated with seven HML-2 proviral loci—HERV-K113 (19p12), HERV-K115 (8p23.1), HERV-K103 (10p12.1), HERV-K106 (3q13.3), HERV-K108 (7p22.1), 3q27.2 (AC069420), and HERV-K107 (5q33.3)—and determined their geographical distribution by amplifying for their presence in 109 DNA samples from four diverse human populations. A schematic diagram of our PCR-based strategy and the predicted outcomes of intraelement homologous recombination are depicted in Fig. 1. Unique 5′ and 3′ flanking region primers were designed in order to detect the preintegration site sequence or solitary LTR at each of the selected loci. The absence of PCR product indicated either the deletion or the presence of a HML-2 provirus. This was evaluated by amplifying for a complete proviral sequence using the unique 5′ flanking primer and universal gag primer. Detection of the allelic variation present at the HERV-K108 loci, which can contain a tandemly repeated provirus (Reus et al. 2001a), initially involved conformational screening for the presence of proviral sequence at that locus, using the unique 5′ flanking region primer and universal gag primer. The presence of at least one copy of the tandemly repeated provirus was analyzed by amplifying with the universal primers gag and env. Computational screening within the human genome databases for the potential combinations of the universal primers gag and env indicated that the predicted amplicon was unique to the HERV-K108 on chromosome 7.
The first polymorphic HML-2 locus that we examined was HERV-K108 on chromosome 7. As we did not perform amplification reactions that spanned the entire length of the HERV-K108 loci, we were unable to distinguish between individuals who were heterozygous in possessing one copy of the ancestral single proviral allele (A) and a copy of the tandemly repeated provirus (B) from individuals who were homozygous for the tandemly repeated provirus (BB) (Table 4). However, in performing conformational amplification for the presence of the HERV-K108 insertion, we were able to determine the number of individuals who were homozygous in possessing the ancestral copy of the provirus (AA). Interelement recombination leading to the production of a tandemly repeated provirus is also expected to generate a solitary LTR. We detected such a solitary LTR in a single individual, indicative of an allele frequency of 0.02 within the African population and a worldwide frequency of 0.005.
The human genomic variation associated with the remaining six HML-2 loci indicated that four of the loci were dimorphic and two loci were monomorphic, consistent with the data retrieved from the human genome databases (Table 5). Allele frequencies for the bi-allelic loci ranged from 0.231 for the HERV-K113 provirus in the Papua New Guinean population to 0.00 for all loci in a number of cases. Interestingly, the allele frequencies for the solitary LTR at the HERV-K103 locus ranged from 0.04 in the African population to zero in all other populations, perhaps suggesting that the solitary LTR has arisen relatively recently. The average heterozygosity values for each locus also varied, from 0.217 for the HERV-K113 locus to 0.00 for the monomorphic loci HERV-K107 and 3q27.2. Only one significant departure from Hardy–Weinberg equilibrium was observed in 24 individual tests; this was for the HERV-K106 solitary LTR in the Papua New Guinean population (data not shown). As 1 of 20 tests are expected to be significant at the 5% level by chance alone, this departure may be due to random statistical fluctuation. The between-population differentiation values for each bi-allelic locus ranged from 0.098 for the HERV-K115 to 0.006 for the HERV-K106 solitary LTR and were all significant by contingency analysis (data not shown). This implies that 90.2 to 99.4% of the genetic variation associated with the polymorphic HML-2 loci is within a population, supporting a recent demographic expansion of contemporary human populations.
Discussion
HERV elements make up a significant proportion of the human genome (8%) and have been proposed to be pacemakers in the evolution of primates (Sverdlov 2000). Determining the structure and cytogenetic location of HML-2 sequences that are unique to humans can be regarded as a starting point for studies investigating their impact, perhaps in regulating the expression of cellular genes or in remodeling the human genome. Here we have reported the structure and cytogenetic location of 74 human-specific HML-2 sequences, of which 15 are complete proviruses and 3 sequences represent near-complete proviral sequences which have lost one of their LTRs (Turner et al. 2001; Barbulescu et al. 1999; Costas 2001; Hughes and Coffin 2001; Sugimoto et al. 2001; Tonjes et al. 1999; Reus et al. 2001a). A single SVA retrotransposon was also characterized, which is located at Xq26.3. Intraelement homologous recombination between the 5′- and the 3′-LTRs of a provirus results in the excision of the retrovirus-like sequence and leaves behind a solitary LTR (Mager and Goodchild 1989). In this study we also describe 49 solitary LTRs, all of which are unique to humans (Mamedov et al. 2002; Medstrand and Mager 1998; Buzdin et al. 2002, 2003; Lebedev et al. 2000; Kurdyukov et al. 2001). A further six sequences have lost the 5′ or 3′ end of their LTR sequence, so we are unable to determine if they were solitary LTRs or complete proviral elements prior to sequence loss. Within this study we have not considered HML-2 sequences which subsist solely as gag, pol, or env genes, although the human genome is likely to contain a significant number of such sequences (Mayer et al. 1997a,b), many of which could be unique to humans.
Copy Number of HML-2 LTRs
The higher proportion of solitary LTRs within the human lineage indicates that the recombination events which led to the loss of structural genes occurred at a faster rate than the retrotransposition, integration, and fixation of novel proviral sequences within the germ line. Further implications are that the solitary LTRs are more genetically stable and/or less deleterious than a full-length provirus and that the recombination events leading to their production are occurring in quick succession after proviral integration. As three of the seven polymorphisms identified within this study (HERV-K103 HERV-K106, and HERV-K108) originate from human-specific proviruses and are generated through intraelement recombination, this observation is confirmed. Interestingly, only 1 individual possessed the HERV-K108 solitary LTR, whereas 46 individuals possessed at least one copy of the reciprocal tandem repeat, perhaps suggesting that the tandem provirus is more genetically stable than the solitary LTR. The increase in HERV-K copy number within the human lineage may also be attributed to complex and recurrent DNA arrangements such as duplication (Medstrand and Mager 1998; Nadezhdin et al. 2001) and is exemplified by the solitary LTR at 6p21.32 (Z80898), which is reported to have arisen through the duplication of the MHC complex (Horton et al. 1998).
Interlocus Recombination
In addition to operating as insertional mutagens, retroelements also serve as substrates for gene conversion and recombination, which has led to a variety of human diseases (Stankiewicz and Lupski 2002; Deininger and Batzer 2002; Ostertag et al. 2003). With the exception of a recombination event between two HERV15 proviruses that flank the AZFa region on the human Y chromosome (Sun et al. 2000; Bosch and Jobling 2003), interelement/interlocus recombination between HERVs is not a frequent cause of human mutation. Despite this, HERV sequences are highly recombingenic (Johnson and Coffin 1999; Hughes and Coffin 2001). The recently recharacterized family of retrotransposons, SVA (SINE, VNTR, and Alu), is derived from SINE and HERV-K(HML-2) elements (Zhu et al. 1994; Ostertag et al. 2003) and a chimeric HERV-H/HERV-K retroelement transposed onto chromosomes 10, 19, and Y before the divergence of the human/chimpanzee gorilla lineages (Lapuk et al. 1999). Recombination or gene conversion has led to the concerted evolution of the HERV-H family (Mager and Freeman 1995) and has also resulted in the homogenization of the LTRs of the RTVL-1a and HERV-K(HML-2) K110/K18 proviral loci (Johnson and Coffin 1999).
Disparate target site duplications are proposed to serve as a signature of involvement of a HERV proviral sequence in interelement/interlocus recombination events. As at least 16% of the HERV-K(HML-2) proviruses that are present within the human genome are estimated to have been involved in such events, they may have had a major impact in primate genome evolution by mediating large-scale chromosomal rearrangements (Hughes and Coffin 2001). We analyzed the direct repeats of all human-specific HML-2 sequences that could be determined to have arisen through the retrotransposition of a HML-2 proviral genome, in order to assess their effect upon the plasticity of the hominid genome. Of the 63 elements that could be considered, two solitary LTRs had disparate target site duplications. PCR amplification and sequencing of their respective preintegration sites in nonhuman primates revealed that neither of these HML-2 loci had been involved in interlocus recombination events. The most parsimonious explanation for the flanking sequence duplication of the solitary LTR at 7p21.2 (Fig. 2A) and the provirus at 21q21.1 (Fig. 2B) is that unequal crossover occurred within a common ancestor who was heterozygous in possessing an allele of the preintegration site sequence and a second allele containing the integrated provirus. If such an event did occur, then the reciprocal sequence would be expected to appear as a preintegration site sequence with a deletion immediately upstream of the site of integration. In the case of the provirus at 21q21.1, in addition to a 450-bp deletion of host chromosomal DNA, the reciprocal would also contain the last 712 bp of the 3′-LTR. We also screened the human genome databases for the expected reciprocal products of unequal crossover and did not detect any such sequences, indicating either that they were not present in the representative individuals sequenced by the human genome projects or that they no longer formed a constituent of the contemporary human gene pool.
Sequence analysis of the preintegration site of the solitary LTR at 17q22 revealed that the downstream direct repeat was 4 bp shorter than the upstream (Fig. 3). The downstream target site duplication could either have lost 4 bp through deletion or during integration when an incomplete target site duplication of only 2 bp was generated. As our results show that disparate direct repeats do not always reflect interlocus recombination events and that at least 3% (2 of 63) of HERV-K(HML-2) sequences, which arose through the retrotransposition of a proviral genome, are a result of unequal crossover, the prediction that at least 16% of HML-2 proviruses have been involved in interlocus recombination events during primate genome evolution may be an overestimate. However, it should be considered that the genomic retroviral elements that exist today represent only a small fraction of total germ line integration and subsequent recombination events that have occurred, namely, those that were not detrimental to the host and that also became fixed in the genome of common ancestors.
Gene Conversion of HML-2 Proviral Loci
Mobile element families are expected to evolve following a “master gene” model of retrotransposition, whereby a few “master” elements give rise to the vast majority of novel sequences with subfamilies evolving either through the accumulation of mutations within the master genes or by the successive replacement of master genes by novel ones. Sequence exchange or gene conversion between different subfamilies of elements can confuse the expected topology, resulting in the apparent accelerated or decelerated evolution of a family (Shih et al. 1991; Mager and Freeman 1995; Kass et al. 1995; Roy-Engel et al. 2002). The phylogeny of HERV-K(HML-2) LTRs presented in this study suggests that a high degree of gene conversion has occurred within the human lineage (Fig. 4). First, the distribution of Type I proviral genomes is not monophyletic, as would be expected if these novel insertions arose from the clonal expansion of a master proviral genome which carried a 292-bp deletion at the pol–env boundary. We also observed similar topology for the gag, pol, and env structural genes (data not shown), indicating that sequence exchange was not restricted to the LTRs. This suggests that the diagnostic 292-bp deletion has been exchanged several times within recent evolutionary time scale between the Type I and the Type II genomes, leading to the production of mosaic proviruses (Costas 2001). Second, the classification of HML-2 LTRs into the subtypes Hs-a and Hs-b (Buzdin et al. 2003) is also not consistent with clonal expansion, as the subtypes group independently of the Type I and Type II proviral genomes. During sequence analysis we also observed that none of the human-specific LTRs contained the diagnostic 8- and 23-bp insertions that are present within the LTRs of the HML-2 ancestor sequences, HERV-K(OLD) (Reus et al. 2001b), indicative of sequence exchange exclusively between highly homologous and recently retrotransposed sequences. The divergent LTRs of the HERV-K115 provirus support this view, as the element is predicted to have entered the human gene pool relatively recently (Turner et al. 2001). However, if gene conversion has occurred as frequently as the Type I/Type II phylogeny suggests, then additional HERV-K(HML-2) proviruses would also be expected to possess highly divergent LTRs. Gene conversion leading to the homogenization of the LTRs within a provirus would counteract this effect and is likely to have occurred regularly within the human lineage as exemplified by the provirus at 12q14.1. This phenomenon has previously been observed within the HERV-K(HML-2) K110/K18 and RTVL-1a proviral loci (Johnson and Coffin 1999). A major consequence of such gene conversion events would be that the accumulative nucleotide differences between the two LTRs of a provirus do not accurately reflect the relative age of the provirus, as demonstrated by the provirus at 11q23.2, which represents an underestimate of time since integration.
HERV-K(HML-2) Polymorphisms for the Study of Human Evolution
HERV insertional or structural mutations leading to the production of a solitary LTR offer several advantages for examining human genomic diversity. First, large numbers of DNA samples can be rapidly typed using PCR-based assays. Second, as with LINE and SINE retroelements, the de novo insertion of a HERV sequence within the germ line represents a unique event in human genome evolution. The large number of potential target sites within the human genome and the random nature of retroviral integration denote that homoplasy is highly unlikely. Third, HERV sequences are stable, as there are no known mechanisms for completely removing them without deleting host chromosomal DNA or leaving behind a solitary LTR. Accordingly, the directionality of the insertion and the formation of a solitary LTR can unambiguously be assigned to a specific lineage, as individual loci containing the same HERV sequence are identical by descent. Fourth, the ancestral state of a HERV sequence is ultimately its absence and is represented by a preintegration site sequence. HERV sequences that are unique to humans can be determined through PCR analysis of the orthologous region in nonhuman primates. This information can be used to root trees of population relationships derived from analysis of HERV polymorphisms. Finally, as the process of reverse transcription generates two LTR sequences that are identical at the time of HERV sequence insertion, the accumulative nucleotide differences between them can serve as a molecular clock (Dangel et al. 1995). However, this measure will be invalidated if a HERV sequence has been subject to recombination or gene conversion after integration.
To ascertain the utility of HERV polymorphisms for examining human evolution, we screened each of the 74 human-specific sequences reported within this study for polymorphism within the human genome databases and determined that seven HML-2 elements were dimorphic. Two of these were solitary LTRs which were polymorphic for insertion. The first is located at 6p21.32 and is reported to have arisen through the duplication of the MHC complex (Horton et al. 1998); the second is located at 9q12. As this chromosomal region is highly repetitive, it is impossible to confirm through PCR amplification the allelic variation of the loci.
In order to examine the genomic variation associated with the remaining five polymorphic HML-2 loci, we developed a PCR-based assay to determine the allelic variation associated with each of them in four diverse human populations. Three bi-allelic loci, HERV-K113, HERV-K115, and HERV-K108, showed geographical distributions that were consistent with previous reports (Reus et al. 2001a; Turner et al. 2001). The two remaining loci, HERV-K103 and HERV-K106, were dimorphic for a solitary LTR and complete copy of the provirus. The HERV-K106 solitary LTR had an average allele frequency of 0.079 and was present in all populations, whereas the HERV-K103 solitary LTR was only present in the heterozygous state in two African individuals. This indicates that the HERV-K103 solitary LTR may be a structural mutation that has arisen relatively recently or that it was unfixed at the time of human expansion from Africa.
Computational screening for the detection of novel retroelement insertion has previously been observed to be subject to bias (Myers et al. 2002). This dictates that high-frequency polymorphisms are lost in the screening process and low-frequency polymorphisms are underrepresented in the human genome databases. We surveyed the genomic variation associated with a further two human-specific HML-2 proviral loci, HERV-K107 and 3q27.2, in order to assess if polymorphism was present. According to the accumulative nucleotide divergence of their respective LTRs, both of these proviruses entered the human gene pool recently and so were likely candidates for insertional or solitary LTR allelic variation. Our results indicated that each of the loci was monomorphic, in accordance with the human genome databases. These results further emphasize that for recent evolutionary events, the accumulative nucleotide differences of the LTRs of a HML-2 provirus do not serve as an accurate measure of time since insertion.
The debate over recent human origins has focused on two models (reviewed by Stringer 2002). The “multiregional model” proposes that over the last 1.5 million years, modern humans arose independently in different regions of the world but remained a single species through worldwide gene flow. In contrast, the “recent replacement model,” or “Out of Africa 2,” suggests that a single population of modern humans migrated from Africa approximately 100,000 to 200,000 years ago and replaced archaic human populations throughout the world. Our survey of the human genomic diversity of HML-2 loci indicates that the genetic diversity of the African population is far higher than non-African populations and that 90.2 to 99.4% of this genetic variability is within a population, supporting a recent demographic expansion of modern humans from Africa.
References
SB Akopov LG Nikolaev PP Khil YB Lebedev ED Sverdlov (1998) ArticleTitleLong terminal repeats of human endogenous retrovirus K family (HERV-K) specifically bind host cell nuclear proteins FEES Lett 421 229–233
M Barbulescu G Turner MI Seaman AS Deinard KK Kidd J Lenz (1999) ArticleTitleMany human endogenous retrovirus K (HERV-K) proviruses are unique to humans Curr Biol 9 861–868
E Bosch MA Jobling (2003) ArticleTitleDuplications of the AZFa region of the human Y chromosome are mediated by homologous recombination between HERVs and are compatible with male fertility Hum Mol Genet 12 341–347
A Buzdin K Khodosevich I Mamedov T Vinogradova Y Lebedev G Hunsmann E Sverdlov (2002) ArticleTitleA technique for genome-wide identification of differences in the interspersed repeats integrations between closely related genomes and its application to detection of human-specific integrations of HERV-K LTRs Genomics 79 413–422
A Buzdin S Ustyugova K Khodosevich I Mamedov Y Lebedev G Hunsmann E Sverdlov (2003) ArticleTitleHuman-specific subfamilies of HERV-K (HML-2) long terminal repeats: three master genes were active simultaneously during branching of hominoid lineages Genomics 81 149–156
FC Chen WH Li (2001) ArticleTitleGenomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees Am J Hum Genet 68 444–456 Occurrence Handle10.1086/318206 Occurrence Handle1:CAS:528:DC%2BD3MXhtl2lsLg%3D Occurrence Handle11170892
J Costas (2001) ArticleTitleEvolutionary dynamics of the human endogenous retro virus family HERV-K inferred from full-length proviral genomes J Mol Evol 53 237–243
AW Dangel BJ Baker AR Mendoza CY Yu (1995) ArticleTitleComplement component C4 gene intron 9 as a phylogenetic marker for primates: Long terminal repeats of the endogenous retrovirus ERV-K(C4) are a molecular clock of evolution Immunogenetics 42 41–52 Occurrence Handle1:CAS:528:DyaK2MXnt1Chsrk%3D Occurrence Handle7797267
PL Deininger MA Batzer (2002) ArticleTitleMammalian retroelements Genome Res 12 1455–1465
AN Domansky EP Kopantzev EV Snezhkov YB Lebedev C Leib-Mosch ED Sverdlov (2000) ArticleTitleSolitary HERV-K LTRs possess bi-directional promoter activity and contain a negative regulatory element in the U5 region FEES Lett 472 191–195
M Faerman D Filon G Kahila CL Greenblatt P Smith A Oppenheim (1995) ArticleTitleSex identification of archaeological human remains based on amplification of the X and Y amelogenin alleles Gene 167 327–332
KA Frazer X Chen DA Hinds PV Pant N Patil DR Cox (2003) ArticleTitleGenomic DNA insertions and deletions occur frequently between humans and nonhuman primates Genome Res 13 341–346
NL Goodchild JD Freeman DL Mager (1995) ArticleTitleSpliced HERV-H endogenous retroviral sequences in human genomic DNA: Evidence for amplification via retrotransposition Virology 206 164–173
R Horton D Niblett S Milne S Palmer B Tubby J Trowsdale S Beck (1998) ArticleTitleLarge-scale sequence comparisons reveal unusually high levels of variation in the HLA-DQB1 locus in the class II region of the human MHC J Mol Biol 282 71–97
JF Hughes JM Coffin (2001) ArticleTitleEvidence for genomic rearrangements mediated by human endogenous retroviruses during primate evolution Nat Genet 29 487–489
JW Huh KW Hong JM Yi TH Kirn O Takenaka WH Lee HS Kim (2003) ArticleTitleMolecular phylogeny and evolution of the human endogenous retrovirus HERV-W LTR family in hominoid primates Mol Cells 15 122–126
InstitutionalAuthorNameInternational Human Genome Sequencing Consortium (2001) ArticleTitleInitial sequencing and analysis of the human genome Nature 409 860–921
WE Johnson JM Coffin (1999) ArticleTitleConstructing primate phylogenies from ancient retrovirus sequences Proc Natl Acad Sci USA 96 10254–10260
DH Kass MA Batzer PL Deninger (1995) ArticleTitleGene conversion as a secondary mechanism in SINE evolution Mol Cell Biol 15 19–25
SG Kurdyukov YB Lebedev II Artamonova TN Gorodentseva AV Batrak IZ Mamedov TL Azhikina SP Legchilina IG Efimenko K Gardiner ED Sverdlov (2001) ArticleTitleFull-sized HERV-K (HML-2) human endogenous retroviral LTR sequences on human chromosome 21: map locations and evolutionary history Gene 273 51–61
AV Lapuk PP Khil IV Lavrentieva YB Lebedev ED Sverdlov (1999) ArticleTitleA human endogenous retrovirus-like (HERV) LTR formed more than 10 million years ago due to an insertion of HERV-H LTR into the 5′ LTR of HERV-K is situated on human chromosomes 10, 19 and Y J Gen Virol 80 835–839
I Lavrentieva P Khil T Vinogradova A Akhmedov A Lapuk O Shakhova Y Lebedev G Monastyrskaya ED Sverdlov (1998) ArticleTitleSubfamilies and nearest-neighbour dendrogram for the LTRs of human endogenous retroviruses HERV-K mapped on human chromosome 19: Physical neighbourhood does not correlate with identity level Hum Genet 102 107–116
YB Lebedev OS Belonovitch NV Zybrova PP Khil SG Kurdyukov TV Vinogradova G Hunsmann ED Sverdlov (2000) ArticleTitleDifferences in HERV-K LTR insertions in orthologous loci of humans and great apes Gene 247 265–277
D Liao T Pavelitz AM Weiner (1998) ArticleTitleCharacterization of a novel class of interspersed LTR elements in primate genomes: Structure, genomic distribution, and evolution J Mol Evol 46 649–660
G Liu S Zhao JA Bailey SC Sahinalp C Alkan E Tuzun ED Green EE Eichler (2003) ArticleTitleAnalysis of primate genomic variation reveals a repeat-driven expansion of the human genome Genome Res 13 358–368
DP Locke R Segraves L Carbone N Archidiacono DG Albertson D Pinkel EE Eichler (2003) ArticleTitleLarge-scale variation among human and great ape genomes determined by array comparative genomic hybridization Genome Res 13 347–357
R Lower J Lower R Kurth (1996) ArticleTitleThe viruses in all of us: Characteristics and biological significance of human endogenous retrovirus sequences Proc Natl Acad Sci USA 93 5177–5184 Occurrence Handle10.1073/pnas.93.11.5177 Occurrence Handle1:STN:280:BymB38visFI%3D Occurrence Handle8643549
DL Mager DJ Freeman (1995) ArticleTitleHERV-H endogenous retroviruses: Presence in the new world branch but amplification in the old world primate lineage Virology 213 395–404
DL Mager NL Goodchild (1989) ArticleTitleHomologous recombination between the LTRs of a human retrovirus-like element causes a 5-kb deletion in two siblings Am J Hum Genet 45 848–854
I Mamedov A Batrak A Buzdin E Arzumanyan Y Lebedev ED Sverdlov (2002) ArticleTitleGenome-wide comparison of differences in the integration sites of interspersed repeats between closely related genomes Nucleic Acids Res 30 e71
J Mayer E Meese N Mueller-Lantzsch (1997a) ArticleTitleChromosomal assignment of human endogenous retrovirus K (HERV-K) env open reading frames Cytogenet Cell Genet 79 157–161
J Mayer E Meese N Mueller-Lantzsch (1997b) ArticleTitleMultiple human endogenous retrovirus (HERV-K) loci with gag open reading frames in the human genome Cytogenet Cell Genet 78 1–5
J Mayer E Meese N Mueller-Lantzsch (1998) ArticleTitleHuman endogenous retrovirus K homologous sequences and their coding capacity in Old World primates J Virol 72 1870–1875
P Medstrand J Blomberg (1993) ArticleTitleCharacterization of novel reverse transcriptase encoding human endogenous retroviral sequences similar to type A and type B retroviruses: Differential transcription in normal human tissues J Virol 67 6778–6787
P Medstrand DL Mager (1998) ArticleTitleHuman-specific integrations of the HERV-K endogenous retrovirus family J Virol 72 9782–9787
JS Myers BJ Vincent H Udall WS Watkins TA Morrish GE Kilroy GD Swergold J Henke L Henke JV Moran LB Jorde MA Batzer (2002) ArticleTitleA comprehensive analysis of recently integrated human Ta L1 elements Am J Hum Genet 71 312–326
EV Nadezhdin YB Lebedev DV Glazkova D Bornholdt IP Arman KH Grzeschik G Hunsmann ED Sverdlov (2001) ArticleTitleIdentification of paralogous HERV-K LTRs on human chromosomes 3, 4, 7 and 11 in regions containing clusters of olfactory receptor genes Mol Genet Genomics 265 820–825
M Ono T Yasunaga T Miyata H Ushikubo (1986) ArticleTitleNucleotide sequence of human endogenous retrovirus genome related to the mouse mammary tumor virus genome J Virol 60 589–598 Occurrence Handle1:CAS:528:DyaL2sXmtF2jtQ%3D%3D Occurrence Handle3021993
EM Ostertag JL Goodier Y Zhang HH Kazazian (2003) ArticleTitleSVA elements are nonautonomous retrotransposons that cause disease in humans Am J Hum Genet 73 1444–1451
C Patience DA Wilkinson RA Weiss (1997) ArticleTitleOur retroviral heritage Trends Genet 13 116–120
K Reus J Mayer M Sauter D Scherer N Muller-Lantzsch E Meese (200la) ArticleTitleGenomic organization of the human endogenous retrovirus HERV-K(HML-2.HOM) (ERVK6) on chromosome 7 Genomics 72 314–320
K Reus J Mayer M Sauter H Zischler N Muller-Lantzsch E Meese (2001b) ArticleTitleHERV-K (OLD): Ancestor sequences of the human endogenous retrovirus family HERV-K (HML-2) J Virol 75 8917–8926
AM Roy-Engel ML Carroll M El-Sawy A Salem RK Garger SV Nguyen PL Deininger MA Batzer (2002) ArticleTitleNon-traditional Alu evolution and primate genomic diversity J Mol Biol 316 1033–1040
W Seifarth C Baust A Murr H Skladny F Krieg-Schneider J Blusch T Werner R Hehlmann C Leib-Mosch (1998) ArticleTitleProviral structure, chromosomal location, and expression of HERV-K- T47D, a novel human endogenous retrovirus derived from T47D particles J Virol 72 8384–8391
L Shen LC Wu S Sanlioglu R Chen AR Mendoza AW Dangel MC Carroll WB Zipf CY Yu (1994) ArticleTitleStructure and genetics of the partially duplicated gene RP located immediately upstream of the complement C4A and C4B genes in the HLA class III region: Molecular cloning, exon-intron structure, composite retroposon, and breakpoint of gene duplication J Biol Chem 269 8466–8476
A Shih EE Coutavas MG Rush (1991) ArticleTitleEvolutionary implications of primate endogenous retroviruses Virology 185 495–502
P Simmonds DB Smith (1999) ArticleTitleStructural constraints on RNA virus evolution J Virol 73 5787–5794
GR Simpson C Patience R Lower RR Tonjes HD Moore RA Weiss MT Boyd (1996) ArticleTitleEndogenous D-type (HERV-K) related sequences are packaged into retroviral particles in the placenta and possess open reading frames for reverse transcriptase Virology 222 451–456
P Stankiewicz JR Lupski (2002) ArticleTitleMolecular-evolutionary mechanisms for genomic disorders Curr Opin Genet Dev 12 312–319
C Stringer (2002) ArticleTitleModern human origins: progress and prospects Philos Trans R Soc Lond B Biol Sci 357 563–579
J Sugimoto N Matsuura Y Kinjo N Takasu T Oda Y Jinno (2001) ArticleTitleTranscriptionally active HERV-K genes: Identification, isolation, and chromosomal mapping Genomics 72 137–144
C Sun H Skaletsky S Rozen J Gromoll E Nieschlag R Oates DC Page (2000) ArticleTitleDeletion of azoospermia factor a (AZFa) region of human Y chromosome caused by recombination between HERV15 proviruses Hum Mol Genet 9 2291–2296
ED Sverdlov (2000) ArticleTitleRetroviruses and primate evolution Bioessays 22 161–171 Occurrence Handle10.1002/(SICI)1521-1878(200002)22:2<161::AID-BIES7>3.0.CO;2-X Occurrence Handle1:CAS:528:DC%2BD3MXmslSht7s%3D Occurrence Handle10655035
RR Tonjes F Czauderna R Kurth (1999) ArticleTitleGenome-wide screening, cloning, chromosomal assignment, and expression of full-length human endogenous retrovirus type K J Virol 73 9187–9195
EM Towler SV Gulnik TN Bhat D Xie E Gustschina TR Sumpter N Robertson C Jones M Sauter N Mueller-Lantzsch C Debouck JW Erickson (1998) ArticleTitleFunctional characterization of the protease of human endogenous retrovirus, K10: Can it complement HIV-1 protease? Biochemistry 37 17137–17144
M Tristem (2000) ArticleTitleIdentification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the human genome mapping project database J Virol 74 3715–3730
G Turner M Barbulescu M Su MI Jensen-Seaman KK Kidd J Lenz (2001) ArticleTitleInsertional polymorphisms of full-length endogenous retroviruses in humans Curr Biol 11 1531–1535
HB Urnovitz WH Murphy (1996) ArticleTitleHuman endogenous retroviruses: nature, occurrence, and clinical implications in human disease Clin Microbiol Rev 9 72–99 Occurrence Handle1:CAS:528:DyaK28XhtFyksLY%3D Occurrence Handle8665478
TV Vinogradova LP Leppik LG Nikolaev SB Akopov AM Kleiman NB Senyuta ED Sverdlov (2001) ArticleTitleSolitary human endogenous retroviruses-K LTRs retain transcriptional activity in vivo, the mode of which is different in different cell types Virology 290 83–90
ZB Zhu B Jian JE Volanakis (1994) ArticleTitleAncestry of SINE-R.C2 a human-specific retroposon Hum Genet 93 545–551
J Zsiros MF Jebbink VV Lukashov PA Voute B Berkhout (1998) ArticleTitleEvolutionary relationships within a subgroup of HERV-K-related human endogenous retroviruses J Gen Virol 79 61–70
J Zsiros MF Jebbink VV Lukashov PA Voute B Berkhout (1999) ArticleTitleBiased nucleotide composition of the genome of HERV-K related endogenous retroviruses and its evolutionary implications J Mol Evol 48 102–111
Acknowledgment
We would like to thank Alastair Macdonald, Rochelle Bleeker, Anna Meredith, Benjamin Searle, and Sonia Lee for their help with sample collection.
Author information
Authors and Affiliations
Corresponding author
Additional information
Reviewing Editor: Dr. Wen-Hsiung Li
Rights and permissions
About this article
Cite this article
Macfarlane, C., Simmonds, P. Allelic Variation of HERV-K(HML-2) Endogenous Retroviral Elements in Human Populations. J Mol Evol 59, 642–656 (2004). https://doi.org/10.1007/s00239-004-2656-1
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-2656-1