
Abstract
Purpose
To assess the potential pharmacokinetic (PK) interactions between siponimod and rifampin, a strong CYP3A4/moderate CYP2C9 inducer, in healthy subjects.
Methods
This was a confirmatory, open-label, multiple-dose two-period study in healthy subjects (aged 18–45 years). In Period 1 (Days 1–12), siponimod was up-titrated from 0.25 to 2 mg over 5 days (Days 1–6) followed by 2 mg once daily on days 7–12. In Period 2, siponimod 2 mg qd was co-administered with rifampin 600 mg qd (Days 13–24). Primary assessments included PK of siponimod (Days 12 and 24; maximum steady-state plasma concentration [Cmax,ss], median time to achieve Cmax,ss [Tmax, ss], and area under the curve at steady state [AUCtau,ss]). Key secondary assessments were PK of M3 and M5 metabolites, and safety/tolerability including absolute lymphocyte count (ALC).
Results
Of the 16 subjects enrolled (age, mean ± standard deviation [SD] 31 ± 8.3 years; men, n = 15), 15 completed the study. In Period 1, siponimod geometric mean Cmax,ss (28.6 ng/mL) was achieved in 4 h (median Tmax,ss; range, 1.58–8.00) and the geometric mean AUCtau,ss was 546 h × ng/mL. In Period 2, the siponimod geometric mean Cmax,ss and AUCtau,ss decreased to 15.7 ng/mL and 235 h × ng/mL, respectively; median Tmax remained unchanged (4 h). Rifampin co-administration increased M3 Cmax,ss by 53% while M5 Cmax,ss remained unchanged. The AUCtau,ss of M3 and M5 decreased by 10% and 37%, respectively. The majority of adverse events reported were mild, with a higher frequency during Period 2 (86.7%) versus Period 1 (50%). The mean ALC increased slightly under rifampin co-administration but remained below 1.0 × 109/L.
Conclusions
The study findings suggest that in the presence of rifampin, a strong CYP3A4/moderate CYP2C9 inducer, siponimod showed significant decrease in Cmax,ss (45%) and AUCtau,ss (57%) in healthy subjects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is a chronic immune-mediated inflammatory disease of the central nervous system (CNS) accompanied by axonal damage, neurodegeneration, and demyelination [1]. The majority of patients (80%) are diagnosed with the relapsing–remitting form (RRMS) of the disease and approximately 50% of these patients eventually develop secondary progressive multiple sclerosis (SPMS) within 10–15 years of disease onset [2].
Siponimod (BAF312) is a potent, oral, and selective modulator of sphingosine 1-phosphate receptor subtypes 1 and 5 (S1P1,5) [3], currently under advanced phase of clinical development for the treatment of SPMS. Siponimod is the first disease modifying therapy showing a robust effect on disability progression in a representative SPMS population [3, 4]. Sphingosine 1-phosphate signalling plays a key role in neuro-inflammatory processes [5, 6]. Siponimod crosses the blood-brain barrier [6]; its interaction with S1P1 limits the inflammatory effects mediated by B cells and T cells [3, 7]. In addition, modulation of S1P receptors is known to limit central inflammation and promote CNS repair in animal models, independent of peripheral immune effects, which may be relevant to SPMS pathology [5, 8,9,10].
In healthy subjects, siponimod has a dose-linear and time-independent pharmacokinetic (PK) profile. The steady state of plasma siponimod is reached after ~ 6 days, with a mean accumulation ratio of 1.9–2.7 and an effective half-life (T1/2) of approximately 30 h (range 22–38 h) [3]. Studies in humans indicate that siponimod is eliminated from the systemic circulation mainly due to metabolism and subsequent biliary/faecal excretion. The polymorphic enzyme CYP2C9 is primarily responsible for the clearance of siponimod (79.2%) with some contribution from CYP3A4 (18.5%) [11]. Metabolite M3 is one of the main circulating metabolites of siponimod in humans [12]; it is formed by glucuronidation of the hydroxylated M5.
Rifampin is a potent activator of the human pregnane X receptor and is classified as a moderate CYP2C9 and a strong CYP3A4 inducer [13]. Moreover, rifampin induces certain drug transporter proteins, such as intestinal and hepatic P-glycoprotein (P-gp) [14]. Rifampin is recommended in regulatory guidelines by the US Food and Drug Administration (FDA) as a prototype inducer to use in human drug–drug interaction (DDI) studies for drugs that are substantially metabolised by CYP2C9/3A4 enzymes [15]. Rifampin is readily absorbed from the gastrointestinal tract. Peak serum concentrations in healthy adults show a wide inter-individual variability. Absorption of rifampin is reduced by approximately 30% when the drug is ingested with food. Rifampin is eliminated via urine and faeces; with repeated administration, the mean T1/2 of rifampin in healthy subjects is 2 to 3 h [15].
Based on the identified CYP enzymes involved in the metabolism of siponimod and in accordance with pertinent regulatory guidelines, the PK, safety, and tolerability of multiple doses of siponimod 2 mg once daily (qd) and its selected metabolites (M3 and M5) were investigated when administered with or without the potent CYP2C9/3A4 inducer rifampin in healthy subjects.
Methods
Subjects
Healthy men and women aged between 18 and 45 years, with a body mass index (BMI) between 18 and 30 kg/m2, total body weight ≥ 50 kg, and a CYP2C9*1*1 (wild type) genotype, were enrolled into the study. Subjects were not eligible for inclusion in the study if they were using any other investigational drugs at the time of enrolment or had a history of hypersensitivity to the study drugs or any contraindication to rifampin. Subjects using any prescription drugs (including vaccines) or herbal supplements within 2 weeks before initial dosing (day 1), and/or over-the-counter medication or dietary supplements (including vitamins) within 1 week before initial dosing, and/or potent inducers of CYP2C9/3A4 within 4 weeks or five half-lives whichever is greater, before initial dosing, and/or potent inhibitors of CYP3A4/2C9 within 5 half-lives before initial dosing, were not enrolled in the study. In addition, subjects with a clinically significant disease in any major system organ class and women of childbearing potential were excluded from the study. Additional subjects were to be enrolled to replace subjects who discontinued the study for reasons other than safety. Except for medication which may be required to treat adverse events, no medication other than study drug was allowed from the first dosing until all of the study completion evaluations were conducted.
Study design
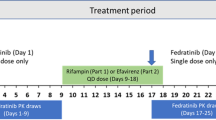
This was a confirmatory, open-label, multiple-dose, two-period, single-sequence study, consisting of a screening epoch (days − 42 to − 2), a baseline period (day − 1), and two treatment epochs (Fig. 1): treatment period 1, where siponimod treatment alone was given on days 1 through 12 (including up-titration) and treatment period 2, where siponimod 2 mg qd was co-administered with rifampin 600 mg qd on days 13 through 24.

Study design. qd, once daily
Siponimod was initiated using the established dose titration regimen. During period 1, subjects received 0.25 mg on the first 2 days, followed by 0.5 mg, 0.75 mg, and 1.25 mg on days 3, 4, and 5, respectively, and a stable dose of 2 mg from day 6 to day 12. Subjects remained at the clinic for the first 6 days for drug administration according to the titration scheme and PK sampling. Subsequently, they continued to visit the clinic for drug administration and any scheduled assessment. During period 2, subjects received concomitantly rifampin 600 mg qd and siponimod 2 mg qd in a fasted state (1 h prior to meals) from days 13 through 24. A study completion evaluation was conducted approximately 7 days (± 2 days) after the last dose of siponimod.
Screening assessments included physical examination, evaluation of medical history, and vital signs, single 12-lead electrocardiogram (ECG), 24-h Holter ECG, and laboratory assessments. Eligible subjects were domiciled at the study site for baseline evaluations approximately 24 h before dosing. After a final assessment of safety and eligibility per protocol, subjects received the study drugs in a fixed sequence of periods 1 and 2.
All study subjects provided written informed consent before any study-specific procedures were performed. The study protocol and subject consent forms were reviewed by an institutional review board. The study was conducted in accordance with the International Council for Harmonisation-Good Clinical Practice Guidelines E6 (R1), with applicable local regulations (including European Directive 2001/20/EC, United States Title 21 Code of Federal Regulations, and Japanese Ministry of Health, Labour, and Welfare) and the ethical principles outlined in the Declaration of Helsinki [16].
Pharmacokinetic assessments
Primary assessments included multiple-dose PK of siponimod alone or in combination with rifampin: maximum steady-state plasma concentration (Cmax,ss), area under the curve at steady state (AUCtau,ss), and median (range) time to achieve Cmax,ss (Tmax,ss). Secondary assessments included multiple-dose PK of the metabolites, M3 and M5, when siponimod was administered alone or in combination with rifampin: Cmax,ss and AUCtau,ss. All PK parameters were calculated using noncompartmental methods with Phoenix® WinNonlin® version 6.4 (Certara USA, Inc., Princeton, NJ, USA).
Safety assessments
Safety assessments included the recording of all adverse events (AEs) and serious AEs (SAEs) with their severity and relationship to the study drug. In addition, safety monitoring also included physical examination, vital signs, clinical laboratory examinations, 12-lead ECG, and 24-h Holter ECG recording (at screening only; no on-treatment Holter ECG recording was performed). Haematology, clinical chemistry, and urinalysis assessments were performed at predefined time points. Absolute lymphocyte count (ALC) levels were assessed separately or as part of the haematology investigations.
Pharmacokinetic blood sampling schedule and bioanalysis
Blood samples were collected by direct venepuncture or an indwelling cannula inserted in a forearm vein. The PK of siponimod and its metabolites M3 and M5 were studied over 24 h on day 12 after multiple administration of siponimod alone (period 1) and on day 24 after co-administration of siponimod and rifampin (period 2). The PK sampling and bioanalysis were conducted per dose on days 1, 2, 3, 4, 5, 6, 8, 10, 12, 13, 15, 17, 19, 21, 22, 23, 24, and 25 (a 24-h post dose sample after the last dose on day 24). Plasma concentrations of siponimod and its metabolites (M3 and M5) were determined by validated liquid chromatography with tandem mass spectrometry methods with a lower limit of quantification of 0.05 ng/mL for siponimod and 0.01 ng/mL for metabolites M3 and M5, respectively. The details of the analytical methods have been described previously [17].
Statistical methods
A total of 16 subjects were recruited to obtain 12 completers in this study. Using the SimCYP physiologically based pharmacokinetics (PBPK) model as described previously [11], siponimod Cmax and AUC were predicted to decrease by 60% and 73%, respectively, when co-administered with rifampin. In order to control the width of confidence intervals (CI) for the geometric mean ratio for siponimod AUC and Cmax in the presence or absence of rifampin, a target was established that the relative half-width was at most 1.25 (0.223 in log scale, 20% from the reference treatment). With the chosen sample size of 12 subjects and an assumed intra-subject coefficient of variation (CV) for AUC of 20% based on previous siponimod studies [18], the relative half-width of 12 subjects is 1.14 (0.131 in log scale, 12% from the reference treatment). The 90% CIs at an observed geometric mean ratio can be evaluated as ratio*exp (− 0.161), ratio*exp (+ 0.161). The screening for the CYP2C9 genotype and exclusion of subjects that were not homozygous for the CYP2C9*1*1 allele (wild type) helped to reduce the inter-subject variability in PK parameters and thereby reduced the minimum sample size required.
The safety analysis set included subjects who received any study drug, whereas the PK analysis set included those subjects who received any study drug with at least one available valid PK concentration measurement, and with no protocol deviations that impacted the PK.
The effect of rifampin on the PK of siponimod and its metabolites in healthy subjects was analysed by using a mixed-effects model. Log-transformed PK parameters Cmax,ss and AUCtau,ss were analysed using a mixed-effects model with treatment as a fixed effect and subject as a random effect. The results were back-transformed to the original scale to obtain adjusted geometric mean ratios and the corresponding 90% CIs for treatment comparison (siponimod + rifampin vs. siponimod alone).
All plasma concentrations that were below the limit of quantification were set to zero for the calculation of the PK parameters. Missing values for any PK parameters were not imputed and were handled as missing values. Descriptive summary statistics were provided for all PK parameters including mean (arithmetic and geometric), standard deviation (SD), CV (arithmetic and geometric), median, minimum, and maximum. Summary tables of treatment-emergent AEs (TEAEs), their severity, and relationship to the study drug, were also provided.
Results
Subject disposition and demographics
A total of 16 subjects were enrolled, and 15 completed the study. One subject was discontinued from the study because of a viral infection during period 1. All 16 subjects (100%) were included in the safety and PK analysis sets. The mean (SD) age of the study population was 31.1 (8.3) years, mean (SD) weight was 79.2 (9.4) kg, and mean (SD) BMI was 25.8 (3.0) kg/m2. The majority of the subjects enrolled were men (n = 15, 94%) with African-American (n = 8, 50%) and Caucasian (n = 6, 37.5%) origin.
Pharmacokinetics of siponimod
Pharmacokinetic parameters of siponimod during both treatment periods have been summarised in Table 1. Following daily oral doses of siponimod, mean trough concentrations reached steady state by day 10. Mean trough concentrations decreased over period 2 and appeared to reach a new steady-state level on day 24 (Fig. 2a).

Arithmetic mean (SD) trough plasma concentration-time profiles of siponimod (a), metabolite M3 (b), and metabolite M5 (c) (PK analysis set). ▲ Period 1: siponimod alone (N = 16). □ Period 2: siponimod + rifampin (N = 15). Period 1: siponimod up-titrated from 0.25 to 2 mg qd (days 1 through 12) (reference). Period 2: siponimod 2 mg qd + rifampin 600 mg qd (days 13 through 24) (test). Values below the limit of quantification (< 0.05 ng/mL) have been set to zero. PK pharmacokinetic, qd once daily, SD standard deviation
After multiple doses of siponimod alone, the geometric mean Cmax,ss was 28.6 ng/mL and the AUCtau,ss was 546 h × ng/mL. The median (range) Tmax,ss was 4 (1.5–8) hours. In the presence of rifampin, the geometric mean Cmax,ss reduced by approximately 45% (15.7 h × ng/mL) while the AUCtau,ss of siponimod decreased by 57% (235 h × ng/mL). The reduced concentrations in period 2 were associated with a higher fluctuation (defined as the degree of fluctuation of the observed minimum concentration to the observed maximum concentration over a dosing interval; 114%) compared with period 1 (56.9%). The median Tmax (4 h) of siponimod was unaltered during both treatment periods. Overall, the variability (CV%) of PK parameters was comparable between the two periods with the exception of the minimum steady-state plasma concentration (Cmin,ss; period 1 CV%: 20.6%; period 2 CV%: 32.9%).
The geometric mean ratios (90% CIs) for siponimod PK parameters indicated that rifampin significantly decreased the exposure parameters of siponimod (Fig. 3a). The ratio of geometric means for Cmax,ss and AUCtau,ss of rifampin co-administered with siponimod versus siponimod alone were 0.55 (0.52; 0.58) and 0.43 (0.41; 0.45), respectively.

Geometric mean ratios (90% CI) for plasma siponimod (a) and its metabolites M3 (b) and M5 (c). Period 1: siponimod alone (reference, day 12); Period 2: siponimod + rifampin (test, day 24). Closed squares and lines represent the geometric mean ratio and 90% CI; open circles represent the individual treatment ratios. AUCtau,ss area under the curve at steady state, CI confidence interval, Cmax,ss maximum steady-state plasma concentration
Pharmacokinetics of M3 and M5
The plasma PK parameters of both M3 and M5 are presented in Table 1. Following daily oral doses of siponimod, the trough concentrations of both M3 and M5 reached steady state by day 10 and decreased over treatment period 2 (Fig. 2b, c).
M3
In the presence of rifampin, the M3 Cmax,ss significantly increased by 53%, with a geometric mean ratio (90% CI) of 1.53 (1.30; 1.81). The AUCtau,ss of M3 decreased by 10%, corresponding to a geometric mean ratio (90% CI) of 0.90 (0.78; 1.03), indicating that the total exposure of M3 remained stable in the absence or presence of rifampin (Fig. 3b). Overall, the variability (CV%) of PK parameters was higher in period 2, but less noticeable for Cmin,ss (period 1 CV%: 37.8%; period 2 CV%: 38.1%). The molecular weight-adjusted M3/parent ratios (MR) for both Cmax,ss and AUCtau,ss increased from day 12 (geometric mean [CV%] 0.422 [51.4%] and 0.388 (47.2%), respectively) to day 24 (1.05 [50.9%] and 0.808 [51.0%], respectively).
M5
The geometric mean ratio of 0.96 (0.86; 1.07) for M5 Cmax,ss was within the default no-effect limits of 0.80 to 1.25, suggesting that the M5 Cmax,ss was stable in the presence of rifampin (Fig. 3c). Rifampin significantly decreased the AUCtau,ss of M5 by 37% with a geometric mean ratio (90% CI) of 0.63 (0.58; 0.68). Overall, the variability (CV%) of PK parameters (AUCtau,ss, Cmax,ss, Cmin,ss, and the average plasma concentration during a dosing interval at steady state Cav,ss) was slightly greater in period 2 than in period 1 (Table 1). The molecular weight-adjusted M5/parent ratios for both Cmax,ss and AUCtau,ss were comparable between day 12 (geometric mean [CV%] 0.0225 [19.5%] and 0.0231 [22.2%], respectively) and day 24 (0.0329 [28.4%] and 0.0392 [22.6%], respectively).
Safety
Overall, 15 of the 16 subjects enrolled (93.8%) experienced at least one TEAE during the study (Table 2). All reported TEAEs, except for one moderate TEAE, were mild in severity and all TEAEs resolved by the end of the study. No serious AEs or deaths were reported during the study. Overall, the most commonly reported TEAEs during the study were chromaturia (n = 8, 50%), dizziness, headache, presyncope, and nausea (reported by three subjects each [18.8%]). The incidence of AEs reported during Period 2 was greater than in Period 1 (eight subjects [50%] vs. 13 subjects [86.7%]). The most common AE reported in Period 1 (three subjects: 18.8% vs. 0% in period 2) was presyncope (two mild and one moderate in severity), mostly following a scheduled blood draw, and none were suspected to be related to study drug. The most common AE in period 2 was chromaturia (eight subjects, 53.3% vs. 0% in period 1), a known side effect of rifampin. One subject was discontinued from the treatment after the drug administration on day 9 (2 mg siponimod) due to a mild viral infection which was suspected to be related to siponimod administration.
Overall, mean values for clinical chemistry parameters and vital signs remained within the normal ranges at all of the assessed time points. There was a mean increase in heart rate (HR) noted after co-administration of siponimod with rifampin, with a maximum value of 73.6 beats per minute (bpm) in day 25 of period 2 (mean change from baseline 10.2 bpm). No clinically significant abnormalities in 12-lead ECG results were observed in the subjects.
Absolute lymphocyte count
A treatment-emergent decrease in ALC was observed in all subjects, representing the desired primary pharmacodynamic effect of siponimod. The magnitude and time course of the mean ALC reduction in period 1 (siponimod alone) was similar to that reported previously during dose titration and at the 2 mg dose level on day 8 [19]. The mean ALC decreased from a baseline of ~ 1.8 × 109/L to ~ 0.8 × 109/L on day 8 (Fig. 4). The mean ALC increased slightly during combination treatment with rifampin in period 2 but remained at levels below 1.0 × 109/L until the completion of period 2 or discontinuation of the study drug; these findings are consistent with the magnitude of steady-state ALC reduction in previous studies at the 1 mg dose level. The mean ALC recovered to near baseline levels (1.73 × 109/L) by the end of the study (approximately 7 days after the last dose).

Absolute lymphocyte count by visit (safety analysis set). The horizontal line in the box interior represents the median. The symbol in the box interior represents the mean. Values outside the whiskers are identified with symbols. The upper (lower) edge of the box represents the 75th (25th) percentile. A whisker is drawn from the upper (lower) edge of the box to the largest (smallest) value within 1.5× interquartile range above (below) the edge of the box. BAS is the last non missing observation before the first study drug administration (including repeated measurements); PER1BAS is the scheduled baseline visit (Study day − 1). BAS baseline, D day, EOS end of study, PER period, qd once daily, SCR screening
Discussion
The present DDI study assessed the effects of the strong CYP3A4/moderate CYP2C9 inducer rifampin on the PK of siponimod and its metabolites in healthy subjects. The simultaneous induction of two siponimod main metabolic pathways, CYP2C9 and CYP3A4, significantly reduced the exposure of siponimod in the presence of rifampin.
An open-label study design was chosen in accordance with the recommendations in the relevant FDA guidelines [15] as it was unlikely that the primary PK endpoint would be influenced by subject or investigator knowledge of the treatment. Healthy subjects were included in the study as no relevant differences are expected in the PK interaction between healthy subjects and MS patients. In order to ensure an accurate quantification of the siponimod (and metabolite) plasma concentration profile over time in case of a potent induction by rifampin, the therapeutic dose of 2 mg of siponimod was used in the study. However, in order to avoid the bradyarrhythmic effect of siponimod at treatment onset, a dose titration over 5 days has been used similar to other siponimod studies [20]. Consequently, this study is an artefactual multiple dose study which is not typically the case with PK-DDI studies with CYP inhibitors or inducers.
Rifampin, as one of the most potent inducers of CYP2C9/3A4, was predicted to significantly decrease the steady-state exposure of siponimod (60% decrease in Cmax and 73% decrease in AUC). Our results showed that in the presence of rifampin, the Cmax,ss and AUCtau,ss of siponimod decreased by 45 and 57%, respectively, which corresponds to the exposure at 1 mg qd steady state. Siponimod trough concentrations decreased over period 2 when rifampin was co-administered and appeared to reach a new steady-state level between day 22 and day 24 in most subjects. This observation confirms that the maximum enzyme induction by rifampin was achieved on day 24 when PK assessments were performed.
The study also measured the PK of two siponimod metabolites, M3 and M5. Metabolite M3 is one of the main circulating metabolites of siponimod in humans [12]; it is formed by glucuronidation of the hydroxylated M5. Metabolite M5 results from metabolism via CYP2C9 mainly, with some contribution from CYP3A4 [17, 21].
They have been toxicologically and pharmacologically profiled in separate studies [12]. M3 and M5 have weak pharmacological activity on S1P1R compared with the parent compound and did not show any signal for toxicity. Metabolite M3 accounts for about 28–39% of the plasma exposure to siponimod, while M5 represents only about 2% of the parent plasma exposure [12].
An increase in M3/M5 exposure is therefore expected when CYP2C9 and CYP3A4 enzymes are induced. Our results indicated that while the Cmax,ss of M3 increased significantly in the presence of rifampin, the AUCtau,ss of M3 displayed only a small change. The M5 Cmax,ss did not change under co-administration of rifampin, but the AUCtau,ss of M5 decreased significantly. Therefore, while induction of CYP2C9 and CYP3A4 significantly increased the clearance of siponimod and increased the peak exposure of M3, rifampin did not increase the total exposure of the M3 and M5 metabolites of siponimod. It has been stated that rifampin also induces the glucuronidation pathway [22]. Since a decrease in M5 exposure was observed, the induced formation of M5 due to the enhanced hydroxylation of siponimod in the presence of rifampin may be counterbalanced by the induced glucuronidation of M5 by rifampin. While the induced glucuronidation is expected to lead to higher systemic levels of the glucuronidated metabolite M3, it was observed that M3 exposure was only marginally modified in the presence of rifampin. Its exposure is likely also influenced by the induction of transporter MRP2 (ABCC2) by rifampin [23], leading to an increase of its biliary excretion and renal elimination. The half-maximal effective concentration (EC50) value of metabolites M5 and M3 tested on human S1P1 receptors were 470 ± 71 nmol/L and > 10,000 nmol/L, respectively. M3 has shown very weak activity on S1P1 (EC50 > 10,000 nmol/L) compared with the parent compound, siponimod, which had shown an EC50 of 1.1 ± 0.41 nmol/L in the same assay [Novartis data on file]. Hence, the observed systemic exposure changes for both M3 and M5 in the presence of rifampin are unlikely to translate into significantly different pharmacological activity on S1P1 receptors.
A recently published study reported that the co-administration of siponimod with fluconazole, a CYP2C9/CYP3A4 inhibitor, resulted in a ~ 2-fold increase in mean AUC versus siponimod alone [24]. In addition, the CYP2C9 genotype has been shown to have a significant impact on siponimod metabolism [11]. The AUCs of siponimod were ~ 2- to-4-fold greater in healthy subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes, with a minor increase in Cmax versus the CYP2C9*1/*1 genotype. The mean T1/2 was prolonged in CYP2C9*2/*3 (51 h) and CYP2C9*3/*3 (126 h) versus CYP2C9*1/*1 (28 h) genotypes [24]. These results further highlighted the relevance of the CYP2C9 pathway on siponimod metabolism.
Beside the induction of a number of drug-metabolising enzymes, rifampin additionally induces some drug transporter proteins, such as P-gp, MRP2, or OATP [14, 25]. In vitro investigations did not identify siponimod as a substrate of efflux transporters (P-gp, BCRP, or MRP2) or of transporters involved in sinusoidal uptake in the liver (OATPs, OCTs, and OATs) (data on file). The regulation of these transporter expression by rifampin is therefore not expected to translate into a clinically relevant changes in siponimod PK.
The CYP2C9 genotype influences the fractional contributions of the two oxidative metabolism pathways (CYP2C9 and CYP3A4) for overall elimination of siponimod: the hepatic CYP2C9 contribution to metabolism is anticipated to be 80.84 and 11.31% in the CYP2C9*1*1 and *3*3 genotypes, respectively, according to physiological-based PK (PBPK) modelling [11]. CYP3A4 plays a minor role in siponimod metabolism for the extensive metaboliser (CYP2C9*1*1 genotype), with a contribution of 17.23% to the overall elimination of siponimod; its contribution to the overall systemic clearance is expected to be greater in subjects with lower CYP2C9 activity associated with a reduced total clearance (78.91% contribution in the *3*3 genotype) [11]. Considering the variable CYP3A4 and CYP2C9 contributions to the overall clearance of siponimod in the different CYP2C9 genotypes, it is expected that the CYP2C9 genotype influences the effects of CYP3A4 and CYP2C9 inhibitors and inducers.
The frequency of the CYP2C9*1/*1 genotype is approximately 62 to 65% in Caucasians [26, 27]. The results of the current study therefore describe the effect of CYP2C9/CYP3A4 induction on siponimod PK in the largest portion of the Caucasian population. As rifampin is a dual (CYP3A4 and CYP2C9) inducer, impacting both siponimod main metabolic pathways, it is expected that the extent of rifampin induction observed in CYP2C9*1/*1 subjects in the current study can be extrapolated to all other CYP2C9 genotypes.
A SimCYP PBPK model has been established using a mixed approach combining in vitro data and PK parameters derived from clinical studies. This model was previously used to accurately predict the effect of fluconazole (a moderate CYP2C9/CYP3A4 inhibitor) co-administration and CYP2C9 genetic polymorphism on siponimod pharmacokinetics in healthy subjects [11, 24]. It is intended to perform complementary PBPK modelling to predict the DDI potential of siponimod as substrate in the presence of typical CYP2C9/3A inducers for the 6 different CYP2C9 genotypes. Overall, multiple oral doses of siponimod alone and in combination with multiple doses of rifampin did not increase the risk of TEAEs in the study. A treatment-emergent decrease in ALC was observed in all subjects, representing the desired primary pharmacodynamic effect of siponimod. The ALC during combination treatment with rifampin was consistent with the magnitude of steady-state ALC reduction in previous studies at the 1-mg dose level [3]. There were no clinically relevant changes in laboratory results, vital signs, and ECG parameters. Minor increases in mean HR were not considered to be related to any study drug in this open-label study.
Conclusion
A clinically relevant decrease in siponimod PK exposure (Cmax,ss and AUCtau,ss) was observed in the presence of rifampin. Multiple oral doses of siponimod alone and in combination with multiple oral doses of rifampin demonstrated a favourable safety profile and were well tolerated by healthy subjects in this study. These results highlight the relevance of the two main metabolic enzymes of siponimod, CYP2C9 and CYP3A4. Considering the expected influence of CYP2C9 genotype on the effects of CYP3A4 and CYP2C9 perpetrator drugs, further evaluations will be required to fully describe the DDI potential of siponimod as substrate in the presence of typical CYP2C9/3A inducers.
Final recommendations on the concomitant use of siponimod and CYP3A4/moderate CYP2C9 inducers including the effect in different genotypes will be made considering all pertinent preclinical, in silico predictions and clinical data.
References
Fitzner D, Simons M (2010) Chronic progressive multiple sclerosis - pathogenesis of neurodegeneration and therapeutic strategies. Curr Neuropharmacol 8(3):305–315. https://doi.org/10.2174/157015910792246218
Scalfari A, Neuhaus A, Degenhardt A, Rice GP, Muraro PA, Daumer M, Ebers GC (2010) The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain 133(Pt 7):1914–1929. https://doi.org/10.1093/brain/awq118
Gergely P, Nuesslein-Hildesheim B, Guerini D, Brinkmann V, Traebert M, Bruns C, Pan S, Gray NS, Hinterding K, Cooke NG, Groenewegen A, Vitaliti A, Sing T, Luttringer O, Yang J, Gardin A, Wang N, Crumb WJ Jr, Saltzman M, Rosenberg M, Wallstrom E (2012) The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br J Pharmacol 167(5):1035–1047. https://doi.org/10.1111/j.1476-5381.2012.02061.x
Kappos L, Bar-Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, Vermersch P, Arnold DL, Arnould S, Scherz T, Wolf C, Wallstrom E, Dahlke F, Investigators EC (2018) Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet 391(10127):1263–1273. https://doi.org/10.1016/S0140-6736(18)30475-6
Brinkmann V (2007) Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther 115(1):84–105. https://doi.org/10.1016/j.pharmthera.2007.04.006
Seabrook T SP SA, Wallström E, Nuesslein-Hildesheim B (2010) Efficacy of the selective S1P1/5 modulator, BAF312 in established EAE and redistribution of S1P1 and S1P5 in the inflamed human CNS tissue. Mult Scler 16(10 Suppl):S301. Abstract P858. In: ed
Amit Bar-Or TD, Vermersch P, Gold R, Tomic D, Cheviet S, Rouyrre N, Scherz T, Hillenbrand R, Kappos L (2017) Longitudinal changes in lymphocyte subsets of siponimod treated patients with secondary progressive multiple sclerosis. Mult Scler 23:P1238 In: ECTRIMSed
Choi JW, Gardell SE, Herr DR, Rivera R, Lee CW, Noguchi K, Teo ST, Yung YC, Lu M, Kennedy G, Chun J (2011) FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A 108(2):751–756. https://doi.org/10.1073/pnas.1014154108
Mullershausen F, Craveiro LM, Shin Y, Cortes-Cros M, Bassilana F, Osinde M, Wishart WL, Guerini D, Thallmair M, Schwab ME, Sivasankaran R, Seuwen K, Dev KK (2007) Phosphorylated FTY720 promotes astrocyte migration through sphingosine-1-phosphate receptors. J Neurochem 102(4):1151–1161. https://doi.org/10.1111/j.1471-4159.2007.4629.x
Gentile A, Musella A, Bullitta S, Fresegna D, De Vito F, Fantozzi R, Piras E, Gargano F, Borsellino G, Battistini L, Schubart A, Mandolesi G, Centonze D (2016) Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J Neuroinflammation 13(1):207. https://doi.org/10.1186/s12974-016-0686-4
Jin Y, Borell H, Gardin A, Ufer M, Huth F, Camenisch G (2018) In vitro studies and in silico predictions of fluconazole and CYP2C9 genetic polymorphism impact on siponimod metabolism and pharmacokinetics. Eur J Clin Pharmacol 74(4):455–464. https://doi.org/10.1007/s00228-017-2404-2
Glaenzel U, Jin Y, Nufer R, Li W, Schroer K, Adam-Stitah S, Peter van Marle S, Legangneux E, Borell H, James AD, Meissner A, Camenisch G, Gardin A (2018) Metabolism and disposition of siponimod, a novel selective S1P1/S1P5 agonist, in healthy volunteers and in vitro identification of human cytochrome P450 enzymes involved in its oxidative metabolism. Drug Metab Dispos 46(7):1001–1013. https://doi.org/10.1124/dmd.117.079574
Cheng J, Ma X, Krausz KW, Idle JR, Gonzalez FJ (2009) Rifampicin-activated human pregnane X receptor and CYP3A4 induction enhance acetaminophen-induced toxicity. Drug Metab Dispos 37(8):1611–1621. https://doi.org/10.1124/dmd.109.027565
Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT (2003) Pharmacokinetic interactions with rifampicin : clinical relevance. Clin Pharmacokinet 42(9):819–850. https://doi.org/10.2165/00003088-200342090-00003
Richter WF, Bhansali SG, Morris ME (2012) Mechanistic determinants of biotherapeutics absorption following SC administration. AAPS J 14(3):559–570. https://doi.org/10.1208/s12248-012-9367-0
Turner-Stokes T, Lu TY, Ehrenstein MR, Giles I, Rahman A, Isenberg DA (2011) The efficacy of repeated treatment with B-cell depletion therapy in systemic lupus erythematosus: an evaluation. Rheumatology (Oxford) 50(8):1401–1408. https://doi.org/10.1093/rheumatology/ker018
Shakeri-Nejad K, Aslanis V, Veldandi UK, Gardin A, Zaehringer A, Dodman A, Su Z, Legangneux E (2017) Pharmacokinetics, safety, and tolerability of siponimod (BAF312) in subjects with different levels of hepatic impairment: a single-dose, open-label, parallel-group study. Int J Clin Pharmacol Ther 55(1):41–53. https://doi.org/10.5414/CP202588
Biswal S, Polus F, Pal P, Veldandi UK, Marbury TC, Perry R, Legangneux E (2015) Pharmacokinetic and pharmacodynamic interaction of siponimod (BAF312) and propranolol in healthy subjects. Int J Clin Pharmacol Ther 53(10):855–865. https://doi.org/10.5414/CP202369
Selmaj K, Li DK, Hartung HP, Hemmer B, Kappos L, Freedman MS, Stuve O, Rieckmann P, Montalban X, Ziemssen T, Auberson LZ, Pohlmann H, Mercier F, Dahlke F, Wallstrom E (2013) Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol 12(8):756–767. https://doi.org/10.1016/S1474-4422(13)70102-9
Legangneux E, Gardin A, Johns D (2013) Dose titration of BAF312 attenuates the initial heart rate reducing effect in healthy subjects. Br J Clin Pharmacol 75(3):831–841. https://doi.org/10.1111/j.1365-2125.2012.04400.x
Gardin A, Dodman A, Kalluri S, Neelakantham S, Tan X, Legangneux E, Shakeri-Nejad K (2017) Pharmacokinetics, safety, and tolerability of siponimod (BAF312) in subjects with severe renal impairment: a single-dose, open-label, parallel-group study. Int J Clin Pharmacol Ther 55(1):54–65. https://doi.org/10.5414/CP202608
Naesens M, Kuypers DR, Streit F, Armstrong VW, Oellerich M, Verbeke K, Vanrenterghem Y (2006) Rifampin induces alterations in mycophenolic acid glucuronidation and elimination: implications for drug exposure in renal allograft recipients. Clin Pharmacol Ther 80(5):509–521. https://doi.org/10.1016/j.clpt.2006.08.002
Fromm MF, Kauffmann HM, Fritz P, Burk O, Kroemer HK, Warzok RW, Eichelbaum M, Siegmund W, Schrenk D (2000) The effect of rifampin treatment on intestinal expression of human MRP transporters. Am J Pathol 157(5):1575–1580. https://doi.org/10.1016/S0002-9440(10)64794-3
Gardin A, Ufer M, Legangneux E, Rossato G, Jin Y, Su Z, Pal P, Li W, Shakeri-Nejad K (2018) Effect of fluconazole co-administration and CYP2C9 genetic polymorphism on siponimod pharmacokinetics in healthy subjects. Clin Pharmacol. https://doi.org/10.1007/s40262-018-0700-3
Jigorel E, Le Vee M, Boursier-Neyret C, Parmentier Y, Fardel O (2006) Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug-sensing receptors in primary human hepatocytes. Drug Metab Dispos 34(10):1756–1763. https://doi.org/10.1124/dmd.106.010033
Scott SA, Khasawneh R, Peter I, Kornreich R, Desnick RJ (2010) Combined CYP2C9, VKORC1 and CYP4F2 frequencies among racial and ethnic groups. Pharmacogenomics 11(6):781–791. https://doi.org/10.2217/pgs.10.49
Kirchheiner J, Brockmoller J (2005) Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther 77(1):1–16. https://doi.org/10.1016/j.clpt.2004.08.009
Acknowledgements
The authors thank subjects and participating centres involved in study. The authors would also like to acknowledge Sivaram Vedantam and Anuja Shah of Novartis Healthcare Pvt. Ltd. Hyderabad, India, for providing medical writing support, which encompassed preparing the manuscript, formatting, referencing, preparing tables and figures, incorporating authors’ revisions, finalising, and submission, all under the direction of the authors. In keeping with the guidelines of the International Committee of Medical Journal Editors, all authors have contributed significantly to the study and were thoroughly involved in the critical review of the manuscript for important intellectual content. All authors have reviewed and approved the final draft for submission.
Funding
The study was funded by Novartis Pharma AG, Basel, Switzerland.
Author information
Authors and Affiliations
Contributions
Anne Gardin and Kasra Shakeri-Nejad conceptualised and designed the studies, analysed and interpreted the study data. Cathy Gray was involved in study design and study execution. Srikanth Neelakantham conceptualised and designed the studies, analysed and interpreted the study data, and conducted statistical analysis of the data. Felix Huth, Antonia M. Davidson, and Eric Legangneux were involved in conceptualising and designing of studies, and acquisition and analysis of data. All authors made significant contribution to data interpretation and review of the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
Antonia M. Davidson is a principal investigator at the PPD Phase 1 Clinic, Austin, TX, USA.
Anne Gardin, Cathy Gray, Srikanth Neelakantham, Felix Huth, Swati Dumitras, Eric Legangneux, and Kasra Shakeri-Nejad are employees of Novartis.
Research involving human participants
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Gardin, A., Gray, C., Neelakantham, S. et al. Siponimod pharmacokinetics, safety, and tolerability in combination with rifampin, a CYP2C9/3A4 inducer, in healthy subjects. Eur J Clin Pharmacol 74, 1593–1604 (2018). https://doi.org/10.1007/s00228-018-2533-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-018-2533-2