
Summary
Pevonedistat (TAK-924/MLN4924) is an investigational small molecule inhibitor of the NEDD8-activating enzyme that has demonstrated clinical activity across solid tumors and hematological malignancies. Here we report the results of a phase 1 study evaluating the effect of rifampin, a strong CYP3A inducer, on the pharmacokinetics (PK) of pevonedistat in patients with advanced solid tumors (NCT03486314). Patients received a single 50 mg/m2 pevonedistat dose via a 1-h infusion on Days 1 (in the absence of rifampin) and 10 (in the presence of rifampin), and daily oral dosing of rifampin 600 mg on Days 3–11. Twenty patients were enrolled and were evaluable for PK and safety. Following a single dose of pevonedistat at 50 mg/m2, the mean terminal half-life of pevonedistat was 5.7 and 7.4 h in the presence and in the absence of rifampin, respectively. The geometric mean AUC0–inf of pevonedistat in the presence of rifampin was 79% of that without rifampin (90% CI: 69.2%–90.2%). The geometric mean Cmax of pevonedistat in the presence of rifampin was similar to that in the absence of rifampin (96.2%; 90% CI: 79.2%–117%). Coadministration of pevonedistat with rifampin, a strong metabolic enzyme inducer, did not result in clinically meaningful decreases in systemic exposures of pevonedistat. The study results support the recommendation that no pevonedistat dose adjustment is needed for patients receiving concomitant CYP3A inducers.
ClinicalTrials.gov identifier
NCT03486314.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pevonedistat (TAK-924/MLN4924) is an investigational small-molecule inhibitor of the neural precursor cell-expressed, developmentally down-regulated 8 (NEDD8)-activating enzyme (NAE) [1, 2]. NAE conjugates NEDD8 to cullin-RING ligases (CRLs), which control ubiquitination and proteasomal degradation of substrates involved in cell cycle progression, DNA replication, oxidative response and response to hypoxia (HIF1a) [3, 4]. The conjugation of NEDD8 to CRLs – neddylation – activates CRLs to ubiquitinate and degrade their substrates [5]. Literature suggests that neddylation has a pivotal role in regulating immune cell function and tumor angiogenesis in the tumor microenvironment [6]. Pevonedistat forms a covalent adduct with NEDD8 that binds to NAE, preventing neddylation, which subsequently leads to apoptotic cell death [7].
Pevonedistat has demonstrated antitumor activities across a variety of tumor cell lines including solid tumors (colon, lung) and hematological malignancies (myeloma, lymphoma) [7,8,9]. The activity of pevonedistat as monotherapy or in combination with standard-of-care agents has been evaluated in multiple tumor types, including advanced solid tumors, melanoma, acute myeloid leukemia, myelodysplastic syndromes, multiple myeloma, and lymphoma [2, 10,11,12,13]. Clinical studies in patients with solid tumors or hematological malignancies evaluated a range of pevonedistat doses and regimens [10,11,12,13]. The maximum tolerated dose (MTD) of pevonedistat was determined to be 25 mg/m2 in combination with docetaxel, 20 mg/m2 in combination with carboplatin plus paclitaxel, and 20 mg/m2 in combination with azacitidine. The recommended dose of pevonedistat for clinical investigation as a single agent was 50 mg/m2 administered on Days 1, 3 and 5 in 21-day cycles.
In a clinical mass balance study, pevonedistat was shown to undergo extensive biotransformation following intravenous administration [14], with preferential distribution in whole blood with a whole-blood-to-plasma ratio of 40. The presence of circulating metabolites, the minor contribution of renal clearance to pevonedistat disposition, and the predominantly urinary and fecal elimination of metabolites suggested that hepatic metabolism plays a major role in the overall clearance of pevonedistat. In a population pharmacokinetics (PK) analysis using data from 335 patients across 6 clinical studies, pevonedistat PKs were linear over the dose range of 25–278 mg/m2 [15]. Age, sex, tumor type, mild or moderate renal impairment (creatinine clearance ≥ 30 mL/min) had no impact on pevonedistat PK. Body surface area (BSA) was identified as a clinically important source of pevonedistat PK variability, supporting the use of BSA-based dosing.
CYP3A was indicated to play a major role in pevonedistat elimination pathways based on in vitro metabolism (Takeda data on file) and the clinical mass balance study [14]. However, co-administration with the strong CYP3A inhibitor itraconazole did not result in clinically meaningful increase in pevonedistat systemic exposure [16]. While strong CYP3A inhibition does not alter pevonedistat PK, the possibility of clinically meaningful reduction in pevonedistat exposure could not be ruled out. There are examples where a relatively minor effect of CYP3A inhibitors may still be consistent with a meaningful reduction in substrate exposure upon strong induction [17, 18]. Here we present results from a study investigating the effects of rifampin, a strong metabolic inducer of pregnane X receptor (PXR)-inducible drug-metabolizing enzymes (e.g., CYP3A) on pevonedistat PK. Since pevonedistat is a cytotoxic agent and cannot be administered to healthy subjects, this study was conducted in patients with advanced solid tumors. Considering concomitant medications are administered to patients with advanced cancer, this study was designed to inform the risk assessment for potential drug-drug interactions with concomitant use of strong CYP3A inducers during the clinical development of pevonedistat.
Methods
Study design
This was an open-label, multi-center study in adult patients with advanced solid tumors (NCT03486314). Eligible patients received a single 50 mg/m2 pevonedistat dose via a 1-h infusion on Days 1 and 10, and daily oral dosing of rifampin 600 mg Days 3–11. Serial blood samples for the measurement of pevonedistat plasma concentrations were collected over 48 h following pevonedistat dosing. Pevonedistat PK parameters and ratios of geometric means for maximal plasma concentration (Cmax) and area under the plasma concentration versus time curve from time 0 to infinity (AUC0–inf) were calculated in the presence of rifampin (Day 10) and in reference to the absence of rifampin (Day 1), with associated 90% confidence intervals (CI) estimated using analysis of variance.
Study objectives
The primary objective was to assess the effect of multiple-dose administration of rifampin on the single-dose PK of pevonedistat in adult patients with advanced solid tumors. The secondary objectives were to further characterize pevonedistat PK, safety and tolerability following a single dose at 50 mg/m2 in the absence or presence of rifampin following a single intravenous dose at 50 mg/m2 in the absence or presence of rifampin.
Patients
Patients aged ≥ 18 years were required to have histologically or cytologically confirmed metastatic or locally advanced and incurable solid tumors that had progressed despite prior standard therapy or for which conventional therapy was not considered effective. Patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and expected survival of at least 3 months. Key exclusion criteria included receiving CYP3A inducers within 2 weeks before the first dose of study drug, treatment with any systemic antineoplastic therapy or any investigational products within 21 days before the first dose of study treatment, or antibiotic therapy, radiotherapy, or major surgery within 14 days. Full eligibility criteria are included in the Supplementary Methods.
Assessments
Serial blood samples for measuring pevonedistat concentrations were collected at intervals from 0 (predose) to 48 h after the single doses of pevonedistat on Day 1 and Day 10. Plasma samples were analyzed for pevonedistat concentrations using a previously reported validated liquid chromatography/mass spectrometry (LC–MS/MS) assay [12].
Safety was assessed by incidence and severity of treatment-emergent adverse events (TEAEs), vital signs, physical examinations, electrocardiograms, and clinical laboratory tests, and toxicities were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.03.
Pharmacokinetic analysis
Pevonedistat PK parameters were calculated by noncompartmental analysis of the pevonedistat plasma concentration–time data using Phoenix WinNonlin version 8.1 (Certara, Princeton, NJ). The following single-dose PK parameters were derived: Cmax, AUC0-inf, AUC from time 0 to the last quantifiable concentration (AUC0–last), terminal disposition phase half-life (t1/2), clearance (CL), and volume of distribution at steady state (Vss).
Statistical analysis
Pevonedistat PK was assessed in all patients who received the protocol-specified pevonedistat and rifampin doses in Part A, and had sufficient concentration–time data to permit reliable estimation of PK parameters (PK-evaluable population). PK data were summarized using descriptive statistics. For assessment of the effect of rifampin on pevonedistat PK, the ratios of geometric mean Cmax, AUClast and AUCinf of pevonedistat in the presence of rifampin on Day 10 versus in the absence of rifampin on Day 1 and associated 90% CI were calculated based on analysis of variance. Estimates for each PK parameter were obtained using a mixed effects model of log(PK parameter) with fixed terms for the rifampin effect and random terms for patient. Safety was assessed in all patients who received at least one dose of pevonedistat, and TEAEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA), version 20.0.
Results
Patients
A total of 20 patients were enrolled in the study. The median age was 63 years, 50% were male, 75% were white, and the mean weight was 87 kg (Table 1). All patients received at least one dose of pevonedistat and completed protocol-specified dosing and PK sampling requirements, thus were included in the safety and PK-evaluable populations.
Pharmacokinetics
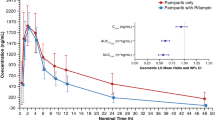
Following a single IV infusion of pevonedistat at 50 mg/m2, the Cmax geometric means were 564.1 and 601.7 ng/mL in the presence and absence of rifampin, respectively. Concentration–time profiles of pevonedistat in the presence and in the absence of rifampin are shown in Fig. 1. The mean terminal t1/2z of pevonedistat was 5.7 h and 7.4 h in the presence and in the absence of rifampin, respectively. Summary statistics of pevonedistat PK parameters are provided in Table 2. The geometric mean AUC∞ of pevonedistat in the presence of rifampin was 79% of the geometric mean AUC∞ of pevonedistat in the absence of rifampin (90% CI: 69.2%–90.2%, N = 20 for Day 1 and N = 17 for Day 10). Similarly, the geometric mean AUClast of pevonedistat in the presence of rifampin was 78.5% of the geometric mean AUClast of pevonedistat in the absence of rifampin (90% CI: 68.4%–90.1%, N = 20 for Day 1 and N = 17 for Day 10). The geometric mean Cmax in the presence and absence of rifampin was similar: 96.2% of the geometric mean Cmax in the absence (90% CI: 79.2%–117.0%, N = 20 for Day 1 and N = 17 for Day 10). Table 2 also presents the statistical analysis of the effect of rifampin on pevonedistat PK. Comparison of individual values of pevonedistat Cmax and AUCs in the presence versus absence of rifampin is illustrated in Fig. 2.

Mean (+ SD) pevonedistat plasma concentration–time profiles in the absence (Day 1) or presence (Day 10) of rifampin (PK population) linear scale (Panel A), semi-log scale (Panel B)

Individual comparisons of AUC∞ and Cmax for pevonedistat in the absence (Day 1) or presence (Day 10) of rifampin (PK population)
Safety
Among 20 patients in the safety population, 16 (80%) patients experienced at least 1 TEAE, and these patients experienced a total of 82 events (Table 3). Nine (45%) patients experienced drug-related TEAEs. Seven (35%) patients experienced grade ≥ 3 TEAEs, and 3 (15%) patients each experienced grade ≥ 3 drug-related TEAEs and serious TEAEs. One patient (5%) died while on study. Two patients experienced a TEAE of hypersensitivity that led to either an interruption of study drug or the drug being permanently withdrawn. The most common TEAEs (≥ 15% of patients) were decreased appetite (4 [20%] patients), fatigue (3 [15%] patients) and diarrhea (3 [15%] patients).
Discussion
Assessment of the effects of intrinsic (e.g., functional capacity of eliminating organs) and extrinsic (e.g., drug–drug interactions) factors on PK plays an essential role in the clinical pharmacology characterization of anticancer agents to inform dosing and administration and risk management in patient populations [19, 20]. Prior to initiating studies to evaluate effects of CYP3A inhibitors or inducers on pevonedistat PK, these concomitant medications were prohibited while patients were receiving pevonedistat in clinical trials. Strong inducers of metabolic enzymes were expected to result in decreased pevonedistat exposures and therefore potential for reduced efficacy.
The selection of the pevonedistat dose of 50 mg/m2 to be investigated in this study was based on the recommended dose for clinical investigation determined in a phase 1 dose-finding for pevonedistat as monotherapy: 50 mg/m2 dosed on Days 1, 3 and 5 in a 21-day treatment cycle. Pevonedistat concentration–time data from 20 patients were used to assess the effect of rifampin, a strong CYP3A inducer, on pevonedistat single-dose PK. Pevonedistat total plasma clearance of 33 L/h and elimination half-life of approximately 7 h in the absence of rifampin were consistent with previously characterized pevonedistat PK [13, 15]. Rifampin produced an approximately 20% decrease in pevonedistat systemic exposure. These results are not consistent with a major contribution of CYP3A-mediated metabolism to the overall clearance of pevonedistat based on in vitro, clinical mass balance and metabolite profiling [14]. Interestingly, itraconazole, a strong CYP3A/P-glycoprotein inhibitor had no clinically relevant effects on pevonedistat PK [16]. To understand the apparent disconnect between in vitro and clinical observations, a physiologically-based pharmacokinetic (PBPK) model for pevonedistat incorporating metabolic clearance and hepatic uptake was developed to explore the mechanisms underlying its lack of sensitivity to clinically observed drug-drug interactions when administered concurrently with CYP3A inhibitors and inducers [21, 22]. The uptake of pevonedistat was assessed using plated human hepatocytes, and the data were analyzed by incorporating the passive diffusion and active transport components in the PBPK model. The model successfully recovered the lack of change in pevonedistat systemic exposures with concomitant treatment of itraconazole, a strong CYP3A inhibitor. This model was also verified by its ability to recover the observation that pevonedistat plasma exposures are not altered by a clinically meaningful extent in the presence of rifampin. The PBPK modeling suggested a significant induction effect on the hepatic metabolism of pevonedistat in the presence of rifampin. However, systemic exposure of pevonedistat was no longer sensitive to the perturbations of enzyme activity if hepatic uptake was the rate-determining step of pevonedistat clearance.
In conclusion, coadministration of pevonedistat with rifampin, a strong metabolic enzyme inducer resulted in an approximately 20% decrease in systemic exposures of pevonedistat. The 20% decrease in systemic exposure of pevonedistat is not considered to be of clinical relevance in the context of an approximately 30% inter-patient variability in pevonedistat clearance [15]. This result supports the recommendation that no pevonedistat dose adjustment is needed when patients are treated with concomitant strong inducers of PXR-inducible enzymes.
Data availability
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participants data supporting the results reported in this article, will be made available within three months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization.
References
Wolenski FS, Fisher CD, Sano T et al (2015) The NAE inhibitor pevonedistat (MLN4924) synergizes with TNF-α to activate apoptosis. Cell death discovery 1:15034. https://doi.org/10.1038/cddiscovery.2015.34
Swords RT, Coutre S, Maris MB et al (2018) Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood 131(13):1415–1424. https://doi.org/10.1182/blood-2017-09-805895
Zhou L, Zhang W, Sun Y, Jia L (2018) Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal 44:92–102. https://doi.org/10.1016/j.cellsig.2018.01.009
Zhou L, Jiang Y, Luo Q, Li L, Jia L (2019) Neddylation: a novel modulator of the tumor microenvironment. Mol Cancer 18(1):77. https://doi.org/10.1186/s12943-019-0979-1
Abidi N, Xirodimas DP (2015) Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr Relat Cancer 22(1):T55-70. https://doi.org/10.1530/erc-14-0315
Jiang Y, Jia L (2015) Neddylation pathway as a novel anti-cancer target: mechanistic investigation and therapeutic implication. Anticancer Agents Med Chem 15(9):1127–1133. https://doi.org/10.2174/1871520615666150305111257
Soucy TA, Smith PG, Rolfe M (2009) Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res 15(12):3912–3916. https://doi.org/10.1158/1078-0432.CCR-09-0343
Milhollen MA, Traore T, Adams-Duffy J et al (2010) MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-(kappa)B-dependent lymphoma. Blood 116(9):1515–1523. https://doi.org/10.1182/blood-2010-03-272567
Swords RT, Kelly KR, Smith PG et al (2010) Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood 115(18):3796–3800. https://doi.org/10.1182/blood-2009-11-254862
Bhatia S, Pavlick AC, Boasberg P et al (2016) A phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with metastatic melanoma. Invest New Drugs 34(4):439–449. https://doi.org/10.1007/s10637-016-0348-5
Lockhart AC, Bauer TM, Aggarwal C et al (2019) Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Invest New Drugs 37(1):87–97. https://doi.org/10.1007/s10637-018-0610-0
Sarantopoulos J, Shapiro GI, Cohen RB et al (2016) Phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with advanced solid tumors. Clin Cancer Res 22(4):847–857. https://doi.org/10.1158/1078-0432.CCR-15-1338
Shah JJ, Jakubowiak AJ, O’Connor OA et al (2016) Phase I study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res 22(1):34–43. https://doi.org/10.1158/1078-0432.CCR-15-1237
Zhou X, Sedarati F, Faller DV et al (2021) Phase I study assessing the mass balance, pharmacokinetics, and excretion of [(14)C]-pevonedistat, a NEDD8-activating enzyme inhibitor in patients with advanced solid tumors. Invest New Drugs 39(2):488–498. https://doi.org/10.1007/s10637-020-01017-x
Faessel HM, Mould DR, Zhou X et al (2019) Population pharmacokinetics of pevonedistat alone or in combination with standard of care in patients with solid tumours or haematological malignancies. Br J Clin Pharmacol 85(11):2568–2579. https://doi.org/10.1111/bcp.14078
Faessel H, Nemunaitis J, Bauer TM et al (2019) Effect of CYP3A inhibitors on the pharmacokinetics of pevonedistat in patients with advanced solid tumours. Br J Clin Pharmacol 85(7):1464–1473. https://doi.org/10.1111/bcp.13915
Cotreau MM, Siebers NM, Miller J, Strahs AL, Slichenmyer W (2015) Effects of ketoconazole or rifampin on the pharmacokinetics of tivozanib hydrochloride, a vascular endothelial growth factor receptor tyrosine kinase inhibitor. Clin Pharmacol Drug Dev 4(2):137–142. https://doi.org/10.1002/cpdd.145
Gupta N, Hanley MJ, Venkatakrishnan K et al (2018) Effects of strong CYP3A inhibition and induction on the pharmacokinetics of ixazomib, an oral proteasome inhibitor: results of drug-drug interaction studies in patients with advanced solid tumors or lymphoma and a physiologically based pharmacokinetic analysis. J Clin Pharmacol 58(2):180–192. https://doi.org/10.1002/jcph.988
Faucette S, Wagh S, Trivedi A, Venkatakrishnan K, Gupta N (2018) Reverse translation of US Food and Drug Administration reviews of oncology new molecular entities approved in 2011–2017: lessons learned for anticancer drug development. Clin Transl Sci 11(2):123–146. https://doi.org/10.1111/cts.12527
Bullock JM, Lin T, Bilic S (2017) Clinical pharmacology tools and evaluations to facilitate comprehensive dose finding in oncology: a continuous risk-benefit approach. J Clin Pharmacol 57(Suppl 10):S105-s115. https://doi.org/10.1002/jcph.908
Varma MV, Lai Y, Feng B et al (2012) Physiologically based modeling of pravastatin transporter-mediated hepatobiliary disposition and drug-drug interactions. Pharm Res 29(10):2860–2873. https://doi.org/10.1007/s11095-012-0792-7
ACCP Abstract Booklet (2021) Clinical Pharmacology in Drug Development. 10(S1):1–104. https://doi.org/10.1002/cpdd.1004
Acknowledgements
The authors thank all the patients who participated in this study and their families, as well as all the investigators and site staff who made the study possible. Editorial support was provided by Ashfield MedComms, an Ashfield Health company, funded by Takeda Pharmaceuticals, U.S.A., Inc., and complied with the Good Publication Practice 3 ethical guidelines (Battisti WP, et al. Ann Intern Med 2015;163:461–4).
Funding
This study was funded by Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Author information
Authors and Affiliations
Contributions
XZ, FS, and KV contributed to the conception of the work. XZ, UV, RDH, KYC, FS, CD, DVF, KV, and NG contributed to the design of the work. UV, DM, RDH, KYC, and FS contributed to data acquisition. XZ, UV, DM, RDH, KYC, FS, and KV contributed to data analysis. All authors contributed to data interpretation, and drafted the work or revised it critically for important intellectual content. All authors provided approval of the final version of the manuscript, and agree to be accountable for all aspects of the work, which includes ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval
This study was conducted in compliance with the independent ethics committee (IEC) requirements of the participating region, informed consent regulations, and applicable regulatory requirements (including International Council for Harmonization [ICH] guidelines), in accordance with ethical principles founded in the Declaration of Helsinki.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Competing interests
XZ is employed by Takeda Development Center Americas, Inc. (TDCA), Lexington, MA, USA. UV has received honoraria from Sanofi, Exelixis, Pfizer, Bayer; consulting fees from BMS, Merck, Exelixis, Bayer, Aveo; and grants/funds from Merck. DM has received honoraria from Amgen, BMS, Exelixis, Eisai; consulting fees from Qurient, OncoOne; and grants/funds from Amgen, Merck, Oncolytics. RDH has received research funding to their institution that supports their salary from: Abbisko, AbbVie, Actuate, Amgen, AstraZeneca, Bayer, Bristol-Myers Squibb, Boston Biomedical, Genmab, GlaxoSmithKline, Infinity, InhibRx, Janssen, Merck, Mersana, Meryx, Nektar, Novartis, Pfizer, Regeneron, Sanofi, Sutro, Takeda, Turning Point Therapeutics, Xencor; and reports consultancy for: Amgen, GlaxoSmithKline. KYC has nothing to disclose. FS is employed by Takeda. CD is employed by Takeda; and has ownership of stock/shares in Takeda and Settle Genetics. DVF is employed by Takeda; has ownership of stock/shares in Viracta Therapeutics, Briacell Therapeutics, Phoenicia Biosciences, R2ccouncil, LLC; was on the advisory council/committee for Viracta Therapeutics, Briacell Therapeutics; and owns multiple patents unrelated to this work. KV is a previous employee of Takeda; and a current employee of EMD Serono Research & Development Institute, Inc., Billerica, MA. NG is employed by Takeda; and has ownership of stock/shares in Takeda.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, X., Vaishampayan, U., Mahalingam, D. et al. Phase 1 study to evaluate the effects of rifampin on pharmacokinetics of pevonedistat, a NEDD8-activating enzyme inhibitor in patients with advanced solid tumors. Invest New Drugs 40, 1042–1050 (2022). https://doi.org/10.1007/s10637-022-01286-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-022-01286-8