
Abstract
Purpose
The aims of this study were to investigate the relationship between metformin exposure, renal clearance (CLR), and apparent non-renal clearance of metformin (CLNR/F) in patients with varying degrees of kidney function and to develop dosing recommendations.
Methods
Plasma and urine samples were collected from three studies consisting of patients with varying degrees of kidney function (creatinine clearance, CLCR; range, 14–112 mL/min). A population pharmacokinetic model was built (NONMEM) in which the oral availability (F) was fixed to 0.55 with an estimated inter-individual variability (IIV). Simulations were performed to estimate AUC0-τ, CLR, and CLNR/F.
Results
The data (66 patients, 327 observations) were best described by a two-compartment model, and CLCR was a covariate for CLR. Mean CLR was 17 L/h (CV 22%) and mean CLNR/F was 1.6 L/h (69%).The median recovery of metformin in urine was 49% (range 19–75%) over a dosage interval. When CLR increased due to improved renal function, AUC0-τ decreased proportionally, while CLNR/F did not change with kidney function. Target doses (mg/day) of metformin can be reached using CLCR/3 × 100 to obtain median AUC0–12 of 18–26 mg/L/h for metformin IR and AUC0–24 of 38–51 mg/L/h for metformin XR, with Cmax < 5 mg/L.
Conclusions
The proposed dosing algorithm can be used to dose metformin in patients with various degrees of kidney function to maintain consistent drug exposure. However, there is still marked IIV and therapeutic drug monitoring of metformin plasma concentrations is recommended.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metformin is the first-line pharmacotherapy in the treatment of type 2 diabetes mellitus (T2DM). Metformin has an excellent safety profile, with favourable properties including weight neutrality and no increased risk of hypoglycaemia. The long-term use of metformin is also associated with a reduction in the risk of diabetes-related deaths [1] and the risk of some cancers [2].
Metformin is cleared by the kidneys, and a dose-proportional reduction with renal function is recommended to reduce the risk of adverse effects such as lactic acidosis [3]. The accumulation of metformin has been previously assessed using peak concentrations of metformin (Cmax < 5 mg/L) [4, 5], and we have proposed dosage regimens of metformin at various stages of renal function to maintain Cmax < 5 mg/L [6].
The pharmacokinetic parameters of immediate release (IR) metformin include a moderate and variable oral availability (55 ± 16% mean ± SD) in healthy subjects [7]. The fractional availability of the extended release (XR) metformin is very similar [8]. There is, however, large inter-individual variability (IIV) in the estimate of the total clearance of metformin (CLTOTAL/F) which is influenced by its oral availability (F). The renal clearance of metformin (CLR), on the other hand, is not affected by F and, therefore, is expected to have a lower IIV than CL/F.
In the present study, we have utilized population pharmacokinetic approaches to investigate the proportion of metformin cleared by the kidneys (CLR), the proportion of the drug not cleared by the kidneys (non-renal clearance of metformin, CLNR/F) and the drug exposure (AUC0–12, AUC0–24) of metformin in a large sample of patients with varying degrees of renal function.
Methods
Patients
Patients receiving metformin (immediate release, IR; extended release, XR) for T2DM were recruited from the outpatient Diabetes Clinic at St Vincent’s Hospital (Australia) and Ziekenhuisgroep Twente Hospital (Almelo, The Netherlands). Demographic characteristics and data on medical comorbidities and concurrent medications were collected. Informed consent was obtained from all individual participants included in the study. Studies 1 and 2 were approved by the Human Research Ethics Committee at St Vincent’s Hospital and University of New South Wales, Sydney (08209/SVH08/035; 09280/SVH09/080), and were registered with the Australian New Zealand Clinical Trials Registry (ACTRN12611000908932). Study 3 was approved by the Ethics Committee of the University Medical Center Groningen (Almelo, The Netherlands; METc 2013.178).
Study design
This was an observational, open-label, cross-sectional study consisting of intensive and sparse blood and urine sampling designs from three studies (studies 1–3, Fig. 1). Blood samples were collected to determine metformin and lactate concentrations in plasma, as well as serum creatinine and HbA1c concentrations. Urine samples were collected to determine metformin and creatinine concentrations. All patients were taking metformin in the long term for the treatment of T2DM.

Study design of study 1, study 2 and study 3. The red dots are the blood sampling time-points, and the grey-shaded area is the urine collection interval (colour figure online)
Study 1
Study 1 was an intensive blood sampling study of patients with a range of kidney function attending the Diabetes Clinic, St Vincent’s Hospital (Sydney, Australia). Information on dosing regimen, dosage and times of last dose prior to blood sampling was collected. All patients were admitted to an observation ward for the study. Patients treated with metformin IR (n = 7) provided blood samples at 0 (pre-dose), 0.5, 1, 2, 3, 4, 6, 8, 10 and 12 h after metformin administration. Urine samples were collected from 0 to 12 h after a regular dose. The blood sampling times for patients treated with metformin XR (n = 9) were 0, 2, 4, 6, 12, 16, 20 and 24 h after their metformin dose. Urine samples for patients treated with metformin XR were collected from 0 to 24 h post dose.
Study 2
Study 2 was a sparse blood sampling study design of patients with chronic kidney disease (CKD; creatinine clearance, CLCR < 40 mL/min) attending the Diabetes Clinic at St Vincent’s Hospital (Sydney, Australia). These patients (n = 5) participated in an interventional study of metformin conducted over 6 weeks [4] and were prescribed daily doses of 500 mg metformin IR. Information on dosing regimen, dosage and times of last dose was collected. A total of eight blood samples and timed urine samples (2–4 h) were collected from each patient. The patients noted the time of their last void, and the 2-h urine sample was collected at the clinic.
Study 3
Study 3 was conducted in patients with mild to moderate kidney disease (n = 45, CLCR < 60 mL/min) attending an outpatient Diabetes Clinic at Ziekenhuisgroep Twente Hospital (Almelo, The Netherlands). Prior to entering the study, patients withheld their usual dose of metformin. The baseline sample was then collected for metformin determination (pre-dose), and patients voided their bladder. After taking their usual dose of metformin, a 2-h blood sample was collected to determine the concentration of metformin in plasma. Urine was collected over 24 h and metformin and creatinine concentrations measured. Patients did not take another dose of metformin during this time interval. The patients collected urine samples at home.
Information on the patients’ dosing regimen was collected; however, the times of last dose were not recorded. Therefore, some assumptions were made when dealing with these data (see “Missing dosage times” section).
Metformin assay
Metformin concentrations in plasma and urine samples were analysed at the Department of Clinical Pharmacology and Toxicology, St Vincent’s Hospital, Australia. For samples collected at Almelo Hospital, plasma and urine samples were stored at −4 °C before transport to St Vincent’s Hospital. The plasma concentrations of metformin were assayed by HPLC using a validated method [6]. Urinary concentrations were assayed similarly.
Population modelling
The plasma concentration data and the accumulated amount of metformin in the urine (mg) were used for population pharmacokinetic analyses. The data were analysed by non-linear mixed effect modelling using NONMEM® version 7.2 (ICON Development Solutions, Ellicott City, MA, USA) with the first-order conditional estimation method with interaction (FOCE-I). For nested models, model selection was informed by using the objective function value (OFV, −2log likelihood), whereby a decrease of >3.84 was considered statistically significant (P < 0.05, χ 2 distribution). The Akaike information criterion (AIC) was used to select the best model between non-nested models. Model runs were executed using Perl-speaks-NONMEM 3.5.3 [9], model development was managed using Pirana 2.8.1 [10] and all plots were generated with R (Version 3.2.0) [11].
Missing dosage times
Because the time of the last dose of metformin was not recorded in study 3, some assumptions regarding the dosing history were necessary for the population pharmacokinetic analyses. Firstly, all subjects using metformin were assumed to be at steady state, receiving metformin IR and compliant with their metformin dosing. Secondly, dosage intervals were assumed to be equal i.e. doses taken every 24 h for once daily dosing, every 12 h for twice daily dosing and every 8 h for thrice daily dosing. Lastly, the patients were asked to withhold their scheduled dose of metformin prior to the start of the study. This was assumed to be the dose of metformin immediately prior to the start of the study. These assumptions were evaluated by investigating the agreement between the observations and the predictions, with and without using the pre-dose time-point for study 3.
Structural and statistical model
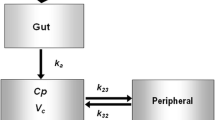
A previously developed population PK model was used [6]. This was a two-compartment model with first-order absorption (k a ) for metformin IR, and zero-order absorption for metformin XR (D) (Fig. 2). The renal clearance of metformin (CLR) was estimated using the accumulated amount of metformin excreted in the urine. Unabsorbed drug together with clearance by all other means is termed the non-renal clearance (CLNR/F).

The final two-compartment model of metformin describing first-order absorption for metformin immediate release (IR) and zero-order absorption for metformin extended release (XR), where k a is the first-order absorption constant and D is the duration of infusion. CLR is the renal clearance, CLNR/F is the non-renal clearance, Q/F is the inter-compartmental clearance, VC/F is the apparent central volume of distribution and VP/F is the apparent peripheral volume of distribution
The following parameters were estimated by the model: CLNR/F, CLR, central volume of distribution (VC/F), inter-compartmental clearance (Q/F) and peripheral volume of distribution (VP/F). The total clearance of metformin (CLTOTAL/F) was calculated using CLR and CLNR/F. From previous studies, the median value of F was 55% [7, 12]. Therefore, F was fixed to a value of 0.55 with an estimated IIV. The IIV for the pharmacokinetic parameters was described using a log-normal distribution:
where P i is the pharmacokinetic parameter of the i th individual, P TV is the typical population parameter value and n i describes the variability between the i th individual and the population parameter which follows a normal distribution with a mean of 0 and a variance of ω 2 i.e. N(0, ω 2).
Several residual error models were tested for metformin concentrations in the plasma and urine. Different error models were tested for each study to account for inaccuracies in sample collection. The combined additive and proportional residual models (mixed error models) were described as
where C ij is the j th measured observation for the i th individual and ε pij and ε aij are the proportional and additive residual random errors, respectively, for the i th individual and the j th measurement. ε pij had a normal distribution of N(0, σ 1 2) and ε aij had a normal distribution of N(0, σ 2 2), where σ 1 and σ 2 represent the standard deviations for the proportional and additive residual error, respectively.
A time-dependent error model [13] was also tested for study 3 due to the missing dosage history. This was investigated as a step function for the plasma concentrations, whereby a different error model was tested for the pre-dose samples, compared to the 2-h post-dose samples.
Covariate model
Covariates for CLR, CLNR/F and VC/F were screened by inspecting scatter plots of empirical Bayes estimates (EBEs) against characteristics of patients (age, sex, weight, lean body weight [14]) and between studies. The equation used for calculating lean body weight was as follows [14]:
where WT is total body weight (kg) and BMI is the body mass index (kg/m2).
Creatinine clearance was tested as a covariate for all clearance parameters and was calculated using the Cockcroft-Gault equation (CLCR) [15] with either total body weight or lean body weight [14, 16]. Creatinine clearance was also calculated directly from the urine output/plasma concentration (UV/P, Eq. 5, CLUCR) and was tested as a covariate for CLR.
Stepwise covariate modelling was utilized to select significant covariates and included the forward selection (OFV ≤3.84 points, P < 0.05, d.f. = 1) and backward elimination of potential covariates (OFV ≤6.63 points, P < 0.01, d.f. = 1).
Model evaluation
The models were evaluated by inspecting diagnostic plots and prediction-corrected visual predictive checks (VPCs, n = 1000). The VPC of the final model was evaluated by comparing the 10th, 50th and 90th percentiles of the observations to the corresponding 10th, 50th and 90th percentiles of the simulations (n = 1000) [17]. A non-parametric bootstrap (n = 1000) stratified by study was used to evaluate the uncertainty of all pharmacokinetic parameter estimates in the final model to obtain the 95% confidence interval for all parameters, as described previously [18].
Dosing simulations
The final model with residual variability and parameter uncertainty was used to simulate 1000 concentration-time profiles at steady-state doses of 500 mg IR and at CLCR of 30, 60, 90 and 120 mL/min. The model-derived AUC0-τ was used to investigate the relationship between drug exposure and CLCR. Relationships between CLTOTAL/F, CLR and CLCR were also investigated.
Results
Patients
A total of 66 patients with T2DM were investigated (Table 1). In study 1, data collection stopped prematurely for two patients taking metformin XR once daily. For these two patients, serial plasma and urine samples were only collected up until 12 and 8 h post dose, respectively. There were no significant differences in the HbA1c values and plasma lactate concentrations between the three studies (Table 1).
Pharmacokinetics
A total of 327 observations from the 66 patients were used for population pharmacokinetic analyses. Over a dosage interval (study 1), the median recovery of metformin in the urine was 49% (19–75%, range) of the dose. The dataset was best described using a two-compartment model with first-order absorption for IR and zero-order absorption for XR. Inter-individual variability (IIV) was added on to the CLNR/F, CLR, VC/F and F parameters, and covariance of CLNR/F and CLR were accounted for in the model. The inclusion of IIV for F reduced the IIV of CLNR/F from 172 to 69%.
A mixed error model was used to describe the residual error for the plasma concentrations of metformin. The model was further improved by using different error models for the urine output for subjects from study 3 (study 1 and study 2 vs study 3, ΔOFV −24.2). A mixed residual error model best described the residual error for the urine concentrations for studies 1 and 2, while an additive residual error best described the residual error for study 3. A time-dependent error model was investigated for study 3, but it did not improve the model predictions.
Despite the lack of dosing information for study 3, there was good agreement between observed and predicted concentrations (without the inclusion of the pre-dose data points) and the eta and epsilon shrinkage was low (<30%), suggesting that the assumptions regarding the times of last doses were valid.
The CLCR using lean body weight was found to be the most significant covariate for CLR and reduced IIV from 45 to 22% (P < 0.01, Eq. 6):
CLCR estimated using Cockcroft-Gault and with lean body weight resulted in a lower OFV than CLUCR. Lean body weight reduced the IIV for VC/F by 4%, but it was not significant at P < 0.01 following backwards elimination of the covariate. Other body size descriptors (total body weight, lean body weight) were not significant covariates for any of the pharmacokinetic parameters. The final model equation for CLR is as follows (Eq. 7):
where θ CLR is the mean population value for CLR, CLCR was centred to the median population value of 2.7 L/h (45 mL/min) and η CLR is the IIV for CLR.
The final model showed good agreement between the observations and the model predictions (Fig. 3) and good agreement between the observations and the model simulations (Fig. 4). Due to the limited number of urine samples collected in the 0- to 12-h post-dose period, wide 95% confidence intervals were observed for the accumulated urine output (Fig. 4). All population pharmacokinetic parameter estimates and their precision are summarized in Table 2. A large residual error was observed for the urine concentrations from study 3, which may be due to the patients’ variable compliance to the study protocol.

Diagnostic plots of the final model. The plots of observations against population predictions and individual predictions were plotted with the line of identity (black) and a linear regression line (blue). Plots of conditional weighted residuals (CWRES) were plotted with a locally weighted scatterplot smoothing curve (red) (colour figure online)

Visual predictive checks (VPCs) of metformin concentrations in plasma (top) and cumulative amount excreted in urine (bottom). The lines represent the 10th, 50th and 90th percentiles of the observations, and the shaded areas represent 95% confidence intervals of the simulated concentrations of the 10th, 50th and 90th percentiles
At metformin doses of 500 mg IR, there was a proportional increase in drug exposure with reduced renal function (Table 3). The CLR of metformin increased approximately in proportion to CLCR. The median ratio of CLR to CLCR was 6.6 (5.1–8.8, 5–95%), and the median ratio of CLTOTAL/F to CLCR was 13 (9–36). The CLNR/F was much smaller than CLTOTAL/F and did not change with renal function (Table 3). Target doses of metformin (while maintaining median Cmax < 5 mg/L, [6]) can be reached using the dosing algorithm (Eq. 8):
This dosing algorithm was used to simulate concentration-time profiles by CLCR (Fig. 5). Since the maximum recommended XR dose of metformin is 2000 mg, this dose was used for dosing simulations at CLCR of 60 to 90 mL/min. Using this dosing algorithm, median AUC0–12 for metformin ranged from 18 to 26 mg/L/h (Table 3). Similarly, the median AUC0–24 for metformin XR ranged from 38 to 51 mg/L/h. Therefore, this dosing algorithm provided a consistent drug exposure across a range of renal function as well as maintaining the median Cmax to be below 5 mg/L (Table 3).

Simulated median plasma concentrations of metformin for CLCR of 15 to 30 mL/min, 60 to 90 mL/min and 90 to 120 mL/min (IR only) at daily doses of 500 mg, 1000 mg, 2000 mg (maximum XR dose) and 3000 mg, respectively
Discussion
It is well accepted that metformin doses should be reduced in patients with impaired renal function because the major mode of elimination is urinary excretion of the unchanged drug.
This study provides further support for a dose-proportional reduction in metformin doses with the decline in kidney function. Based on the recommendation to keep metformin Cmax below 5 mg/L [5, 6], we have proposed a dosing algorithm to estimate appropriate doses by renal function. The maximum recommended daily dose of metformin is 3000 mg for IR and 2000 mg for XR [19]. In our simulations, daily doses of 2000 mg XR were used for CLCR of 60 to 90 mL/min (XR only). If higher doses are required, patients should be switched from metformin XR to metformin IR, provided that they do not experience gastrointestinal side-effects [19]. For metformin IR, median AUC0–12 was 18 to 26 mg/L/h, which is similar to AUC0–24 for metformin XR (38 to 51 mg/L/h) for patients with CLCR from 15 to 90 mL/min. However, the ranges of peak plasma concentrations and AUC values are very wide (Table 3) and we therefore suggest that the plasma concentrations should be measured in order to optimize dosage with this important drug.
This is the first population pharmacokinetic model of metformin to describe the relationship between CLR, CLNR/F and drug exposure in patients with T2DM and kidney disease, and CLCR estimated using Cockcroft-Gault and with lean body weight was a significant covariate for CLR. Unlike our previous model [6], body weight was not a covariate for VC/F. This is likely due to the high shrinkage for VC/F (37%), which may have hidden the true covariate relationship with weight [20]. The median value of F in our study was 49 with (range, 19 to 75%). This result was similar to the values of 55 ± 19% (mean ± SD) [12].
There are limited and conflicting results on the non-renal clearance of metformin. Previous studies on the urinary recovery of metformin after the reference intravenous injection have reported either complete recovery [21] or 80% recovery in the urine, with no metformin recovered in the faeces [12]. By comparison, our pharmacokinetic analysis indicates that the CLNR/F is much smaller than CLR. Further studies are required to investigate whether a non-renal elimination pathway exists for metformin in man. In rats, the metabolism of metformin was suggested due to the reduced half-life of metformin by cytochrome P450 enzymes and lengthened by inhibitors of these enzymes, but no corresponding study has been conducted in man [22].
The ratio of CLR and CLTOTAL/F with CLCR were higher than estimated previously. In our review of the pharmacokinetics of metformin, the ratio of CLR to CLCR was 4.0 ± 1.5 [7] compared to a median value of 6.6 (5 to 95% range 5.1 to 8.8) in the present studies. Further, the ratio of the CLTOTAL/F in our review was 10.7 ± 3.5 which is considerably lower than the ratio 13.1 (5 to 95% range 9.3 to 36.5) in the present studies. The reasons for the contrast are unclear, but many patients included in our previous review either did not have T2DM or were administered single doses of metformin. By contrast, all patients in the present study were dosed with metformin in the long term for their T2DM. Furthermore, the wide range of CLTOTAL/CLCR in the present study is probably due to the large inter-patient variation of F.
There were some limitations to the present study. The shrinkage on VC/F was high due to the sparse collection of pharmacokinetic time-points for studies 2 and 3, which may have masked the true covariate effect of weight for VC/F. Additionally, there were a limited number of urine samples collected over the dosage interval and all urine collections were assumed to be complete. Study 1 was conducted entirely in the research centre while patients enrolled in study 2 were required to report the times of last void and patients from study 3 collected their 24-h urine at home. Poor compliance to the study protocol would contribute to the variability in the estimate of urinary recovery.
Potential compliance issues for study 3were accounted for using a separate residual error model for the urine concentrations. This error model revealed a large additive residual error for study 3, which significantly improved the model and indicated poor compliance to urine collection at home.
Conclusion
We have described the CLR, CLNR and AUC of metformin in patients with varying degrees of renal function. We have proposed a dosing algorithm that can be used to reduce metformin doses proportionally with kidney function to maintain Cmax below 5 mg/L and to achieve a consistent drug across a range of renal function. CLR was well estimated; however, further studies are required to obtain reliable estimates of CLNR/F and F.
References
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA (2008) 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359(15):1577–1589. doi:10.1056/NEJMoa0806470
Chen YC, Kok VC, Chien CH, Horng JT, Tsai JJ (2015) Cancer risk in patients aged 30 years and above with type 2 diabetes receiving antidiabetic monotherapy: a cohort study using metformin as the comparator. Ther Clin Risk Manag 11:1315–1323. doi:10.2147/TCRM.S91513
Duong JK, Furlong TJ, Roberts DM, Graham GG, Greenfield JR, Williams KM, Day RO (2013) The role of metformin in metformin-associated lactic acidosis (MALA): case series and formulation of a model of pathogenesis. Drug Saf. doi:10.1007/s40264-013-0038-6
Duong JK, Roberts DM, Furlong TJ, Kumar SS, Greenfield JR, Kirkpatrick CM, Graham GG, Williams KM, Day RO (2012) Metformin therapy in patients with chronic kidney disease. Diabetes Obes Metab. doi:10.1111/j.1463-1326.2012.01617.x
Lalau JD, Lemaire-Hurtel AS, Lacroix C (2011) Establishment of a database of metformin plasma concentrations and erythrocyte levels in normal and emergency situations. Clin Drug Investig 31(6):435–438. doi:10.2165/11588310-000000000-00000
Duong JK, Kumar SS, Kirkpatrick CM, Greenup LC, Arora M, Lee TC, Timmins P, Graham GG, Furlong TJ, Greenfield JR, Williams KM, Day RO (2013) Population pharmacokinetics of metformin in healthy subjects and patients with type 2 diabetes mellitus: simulation of doses according to renal function. Clin Pharmacokinet 52(5):373–384. doi:10.1007/s40262-013-0046-9
Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, Furlong TJ, Greenfield JR, Greenup LC, Kirkpatrick CM, Ray JE, Timmins P, Williams KM (2011) Clinical pharmacokinetics of metformin. Clin Pharmacokinet 50(2):81–98. doi:10.2165/11534750-000000000-00000
Timmins P, Donahue S, Meeker J, Marathe P (2005) Steady-state pharmacokinetics of a novel extended-release metformin formulation. Clin Pharmacokinet 44(7):721–729. doi:10.2165/00003088-200544070-00004
Lindbom L, Pihlgren P, Jonsson EN (2005) PsN-toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Prog Biomed 79(3):241–257. doi:10.1016/j.cmpb.2005.04.005
Keizer RJ, van Benten M, Beijnen JH, Schellens JH, Huitema AD (2011) Pirana and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Prog Biomed 101(1):72–79. doi:10.1016/j.cmpb.2010.04.018
R Core Team (2015). R: a language and environment for statistical computing. R foundation for statistical computing, Vienna. URL http://www.R-project.org/.
Tucker GT, Casey C, Phillips PJ, Connor H, Ward JD, Woods HF (1981) Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol 12(2):235–246
Karlsson MO, Beal SL, Sheiner LB (1995) Three new residual error models for population PK/PD analyses. J Pharmacokinet Biopharm 23(6):651–672
Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B (2005) Quantification of lean bodyweight. Clin Pharmacokinet 44(10):1051–1065. doi:10.2165/00003088-200544100-00004
Cockcroft DW, Gault MH (1976) Prediction of creatinine clearance from serum creatinine. Nephron 16(1):31–41
Janmahasatian S, Duffull SB, Chagnac A, Kirkpatrick CM, Green B (2008) Lean body mass normalizes the effect of obesity on renal function. Br J Clin Pharmacol 65(6):964–965. doi:10.1111/j.1365-2125.2008.03112.x
Karlsson MO, Holford N (2008) A tutorial on visual predictive checks. PAGE. Abstr 1434 [www.page-meeting.org/?abstract=1434]
Henderson AR (2005) The bootstrap: a technique for data-driven statistics. Using computer-intensive analyses to explore experimental data. Clin Chim Acta 359(1–2):1–26. doi:10.1016/j.cccn.2005.04.002
Australian Medicines Handbook (2017) Adelaide: Australian Medicines Handbook Pty Ltd
Savic RM, Karlsson MO (2009) Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J 11(3):558–569. doi:10.1208/s12248-009-9133-0
Pentikainen PJ, Neuvonen PJ, Penttila A (1979) Pharmacokinetics of metformin after intravenous and oral administration to man. Eur J Clin Pharmacol 16(3):195–202
Choi YH, Lee MG (2006) Effects of enzyme inducers and inhibitors on the pharmacokinetics of metformin in rats: involvement of CYP2C11, 2D1 and 3A1/2 for the metabolism of metformin. Br J Pharmacol 149(4):424–430. doi:10.1038/sj.bjp.0706875
Acknowledgements
This work was performed using computing infrastructure provided by the Australian Centre of Pharmacometrics. The Australian Centre for Pharmacometrics is an initiative of the Australian Government as part of the National Collaborative Research Infrastructure Strategy.
Author information
Authors and Affiliations
Contributions
JKD designed and conducted studies 1 and 2, analysed the data, built the model and wrote the manuscript. MK conducted study 3, analysed the data and contributed to the manuscript. SSK and CMK provided advice on the population model and interpretation of the results. GG was involved in detailed discussions on the clinical significance of the results with JKD. All authors reviewed the manuscript and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Informed consent was obtained from all individual participants included in the study. Studies 1 and 2 were approved by the Human Research Ethics Committee at St. Vincent’s Hospital and University of New South Wales, Sydney (08209/SVH08/035; 09280/SVH09/080), and were registered with the Australian New Zealand Clinical Trials Registry (ACTRN12611000908932). Study 3 was approved by the Ethics Committee of the University Medical Center Groningen (Almelo, The Netherlands; METc 2013.178).
Funding
This study was funded by the NH&MRC Program Grant 568612, Australian Research Council Grant LP 0990670 and St Vincent’s Clinic Foundation Sister Mary Bernice Research Grant. JKD and SSK had support from the Australian Research Council (ARC) Linkage Grant LP 0990670 for the submitted work.
Competing interests
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Duong, J.K., Kroonen, M.Y.A.M., Kumar, S.S. et al. A dosing algorithm for metformin based on the relationships between exposure and renal clearance of metformin in patients with varying degrees of kidney function. Eur J Clin Pharmacol 73, 981–990 (2017). https://doi.org/10.1007/s00228-017-2251-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-017-2251-1