
Abstract
Purpose
The aim of the review was to provide an overview of studies investigating the pharmacokinetics of exogenous melatonin in humans and if possible, to provide recommendations for clinical use.
Methods
The review was conducted in accordance to PRISMA guidelines. A systematic literature search was performed in PubMed and Embase databases. The pharmacokinetic variables included maximal plasma/serum concentration (Cmax), time to maximal plasma/serum concentration (Tmax), elimination half-life (T1/2), area-under-the-curve plasma/serum concentrations (AUC), clearance (Cl), volume of distribution (VD), and bioavailability.
Results
The literature search identified 392 records. Twenty-two studies were included in the review. Melatonin dosages varied between 0.3 and 100 mg and were administered either orally or intravenously. Cmax ranged from 72.1 (10 ml/h; 0.02 mg, IV) to 101,163 pg/ml (100 mg, oral). Tmax ranged between 15 (2 mg) and 210 min (10 mg). T1/2 ranged from 28 (0.005 mg, IV) to 126 min (4 mg, oral), whereas AUC ranged between 5400 (0.005 mg, IV) and 6.56 × 1010 pg/ml × min (1 mg, oral). Cl ranged from 0.97 (0.005 mg, IV) to 132.50 L/min (6 mg, oral), whereas VD ranged between 35 (0.005 mg, IV) and 1602 L (4 mg, oral). Bioavailability of oral melatonin ranged from 9 to 33 %. Pharmacokinetics was affected by age, caffeine, smoking, oral contraceptives, feeding status, and fluvoxamine. Critically ill patients displayed accelerated absorption and compromised elimination.
Conclusions
Despite methodological differences between the included studies, Tmax was approximately 50 min following oral immediate-release formulations of melatonin. T1/2 was 45 min in both administration routes. Cmax, AUC, Cl, and VD varied extensively between studies. Bioavailability of oral melatonin was approximately 15 %.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The pineal hormone, melatonin regulates a variety of physiological functions, such as circadian rhythm, reproduction, mood, and immune function [1]. Correspondingly, exogenous melatonin has demonstrated a number of clinical effects [1, 2]. Numerous clinical studies have documented improved sleep quality following administration of exogenous melatonin [1, 3, 4]. Recent studies also demonstrate analgesic [4, 5], anxiolytic [5], anti-inflammatory [6], and anti-oxidative effects [6, 7] of melatonin.
In clinical studies, melatonin is typically administered orally, sublingually, or intravenously. Until now, the pharmacokinetics of melatonin has primarily been investigated in healthy volunteers following oral and intravenous administration of melatonin, but findings have been inconsistent [8, 9]. The increasing clinical use, however, necessitates further investigation of the pharmacokinetics of melatonin, and an overview of the current literature is urgently needed.
The aim of this systematic review was to provide a comprehensive overview of studies investigating pharmacokinetics of exogenous melatonin in humans and if possible, to provide evidence-based recommendations for clinical use.
Methods
The review was performed in accordance to PRISMA guidelines [10] and registered at PROSPERO (CRD42014015040). A systematic literature search was conducted in PubMed and Embase databases in October 2014. The search strategy included following search algorithm: Melatonin AND (exogenous OR exogenously OR administration) AND (pharmacokinetic OR pharmacokinetics OR bioavailability). Furthermore, a manual snowball search was performed in the reference lists of the included studies. Two independent authors (NGH, LPHA) performed the literature search. If any disagreements occurred between the authors, consensus was achieved between all authors of the review.
The review included all studies written in English investigating pre-defined pharmacokinetic variables following exogenous administration of melatonin in adult volunteers and patients. Studies, which did not fulfill these criteria or investigated melatonin receptor agonists, were excluded from the review.
The pre-defined pharmacokinetic variables included the following: maximal plasma/serum concentration (Cmax), time to maximal plasma/serum concentration (Tmax), elimination half-life (T1/2), area-under-the-curve plasma/serum concentrations (AUC), clearance (Cl), volume of distribution (VD), and bioavailability (units: pg/ml, min, min, pg/ml × min, L/min, L, and %, respectively). If the pharmacokinetic variables appeared in different units than described above, relevant adjustments were performed. Body weight (BW)-adjustments of data were employed, if needed, applying a correction value of 75 kg BW. For oral administrations of melatonin, bioavailability-corrected values of Cl and VD were employed and reported as Cl/F and VD/F. Additional outcomes reported in the included studies, apart from the pre-defined pharmacokinetic variables, are not presented or evaluated in this review.
Results
The complete literature search is described in detail in Fig. 1. Twenty-two studies (n = 359) were included in the review [3, 8, 9, 11–29].

PRISMA flowchart
Table 1 describes number of volunteers/patients, study design, volunteer/patient characteristics, measuring period, and reported pharmacokinetic variables.
The review included five randomized clinical trials (RCT) [3, 8, 17, 21, 28], 16 cohort studies [9, 11–16, 18–20, 22, 23, 25–27, 29], and one case report [24]. Nineteen studies included healthy volunteers [8, 9, 11–15, 17–20, 22–29]. Three clinical studies included critically ill patients [3, 16] and elderly patients suffering from insomnia [21], respectively.
Four studies administered melatonin by intravenous bolus injection [8, 12, 18, 24] in doses ranging from 0.0005 mg/kg to 2 mg [8, 12]. Two studies applied intravenous infusions in doses of 0.02 mg (10 ml/h) and 0.023 mg (250 ml/h) [18, 20], respectively. Eighteen studies administered oral melatonin (capsules: n = 8 [11, 13, 15, 19, 25–28]/tablets: n = 7 [16, 17, 21–23, 25, 29]/oral solutions: n = 3 [3, 20, 25]/powders: n = 1 [27]) in doses ranging from 0.3 [11] to 100 mg [13]. The studies investigating oral melatonin administered immediate-release formulations in 17 studies [3, 8, 9, 11, 13–17, 19, 20, 22, 23, 25, 27–29], surge-sustained release (25 % immediate-release + 75 % controlled release) formulations in one study [21], pulsatile-release (immediate-release+secondary release) formulations in one study [28], and slow-release formulations in two studies [25, 26]. All oral administrations were single doses, except for one study administering three separate doses [15]. The measuring periods ranged from 2 to 36 h [15, 18]. Plasma/serum melatonin concentrations were assessed by radioimmunoassay (RIA) in 15 studies [3, 8, 11–13, 15–19, 21, 24, 25, 28, 29], by high-performance liquid chromatography (HPLC) in three studies [22, 23, 26] and by gas chromatography-mass spectrometry (GC-MS) in three studies [14, 20, 27]. One study did not report the applied assay technique [9].
Pharmacokinetic variables
The reported pharmacokinetic variables Cmax, Tmax, T1/2, AUC, Cl, VD, and bioavailability of each study are presented in Table 2.
Cmax was measured in 18 studies [3, 8, 11, 13, 14, 16–23, 25–29]. Cmax after intravenous bolus administration was 96,850 pg/ml (dose 2 mg) [8]. For intravenous infusions, Cmax ranged between 72.1 pg/ml (dose 0.02 mg, 10 ml/h) [18] and 169.0 pg/ml (dose 0.023 mg, 250 ml/h) [20]. After oral administrations, Cmax ranged from 170 (dose 0.3 mg) [11] to 101,163 pg/ml (dose 100 mg) [13].
Tmax was reported in 17 studies [3, 11, 13–17, 19–28]. For intravenous infusions, Tmax was 113.4 min for male volunteers and 110.4 min for female volunteers (dose 0.023 mg, 250 ml/h), respectively [20]. Tmax for oral immediate-release formulations ranged from 15 (dose: 2 mg) [25] to 90 min (dose 25 mg) [19]. The oral surge-sustained formulations displayed Tmax values of 78 and 90 min, respectively (dose 0.4 and 4 mg) [21]. Two different oral pulsatile-release formulations displayed Tmax of 45 and 210 min, respectively (dose 10 mg) [28]. One study employing an oral slow-release formulation reported a Tmax value of 167 min (dose 5 mg) [26].
T1/2 was assessed in 18 studies [3, 8, 9, 12, 13, 15–18, 20–26, 28, 29]. After intravenous bolus administrations, T1/2 ranged from 28 (dose 0.005 mg) [18] to 60 min (dose 2 mg) [8]. After intravenous infusions, T1/2 ranged between 36 (dose 0.023 mg, 250 ml/h) [20] and 45 min (dose 0.02 mg, 10 ml/h) [18]. Following oral administrations, T1/2 ranged from 32 (dose 2 mg) [25] to 126 min (dose 4 mg) [21].
AUC was reported in 17 studies [3, 8, 11, 12, 15, 16, 18–23, 25–29]. AUC after intravenous bolus administrations ranged from 5400 pg/ml × min (dose 0.005 mg) [18] to 1.63 × 106 pg/ml × min (dose 2 mg) [8]. AUC for intravenous infusions was 15,288 pg/ml × min for male volunteers and 21,846 pg/ml × min for female volunteers (dose 0.023 mg, 250 ml/h), respectively [20]. Following oral administrations, AUC ranged between 6.56 × 1010 pg/ml × min (dose 1 mg) [27] and 14,160 pg/ml × min (dose 0.25 mg) [20]. One study, applying a multiple dosage regimen documented an AUC of 31.3 × 106 (dose 80 mg × 3 per hour) [15].
Cl was reported in nine studies [3, 12, 17, 18, 20–23, 26]. Cl after intravenous bolus administrations ranged from 0.97 (dose 0.005 mg) [18] to 3.30 L/min (dose: 0.0005 mg/kg) [12]. After intravenous infusions, Cl ranged between 0.97 (dose 0.02 mg, 10 ml/h) [18] and 1.57 L/min (dose 0.023 mg, 250 ml/h) [20]. Following oral administrations, Cl/F ranged from 1.63 (dose 6 mg) [17] to 132.50 L/min (dose 6 mg) [23].
VD was reported in five studies [12, 18, 20, 21, 26]. Following intravenous bolus administration, VD ranged from 35 (dose 0.005 mg) [18] to 185 L (dose 0.0005 mg/kg) [12]. After intravenous infusions, VD values ranged between 53.8 and 73.1 L (dose 0.023 mg, 250 ml/h) [20]. Following oral administrations, VD/F ranged from 451 (dose 5 mg) [26] to 1602 L (dose 4 mg) [21].
Three studies reported absolute bioavailability [8, 9, 20]. Bioavailability ranged from 9 (dose 0.25 mg) [20] to 33 % (dose 0.5 mg) [9].
Internal factors
A cohort study including 36 volunteers investigated the effect of age [11]. Volunteers were grouped as either young (20–43 years) or older (49–73 years) subjects. The study did not document any significant differences in Cmax, AUC or Tmax values between the groups [11].
The effect of age and gender was also investigated in another cohort study including 33 volunteers (6–31 years) [12]. Prepubertal volunteers (mean age = 8.4 years) displayed significantly shorter T1/2 and a lower AUC compared to adult volunteers (mean age = 27.2 years). No difference between the genders was observed.
The effect of menopause was investigated in a cohort study including 18 female volunteers [22]. The pharmacokinetic variables Cmax, Tmax, T1/2, AUC, and Cl/F of melatonin were not significantly different between premenopausal and postmenopausal women [22].
External factors
The effect of caffeine, cigarette smoking, and genotype on pharmacokinetic variables of melatonin was investigated in a RCT including 12 volunteers [17]. The volunteers were randomized to either melatonin (6 mg) or melatonin (6 mg) + caffeine (3 × 200 mg). Concomitant administration of caffeine increased Cmax from 4480 to 10,618 pg/ml, increased T1/2 from 106 to 113 min, and reduced Cl/F from 3.08 to 1.63 L/min compared to melatonin alone [17]. Furthermore, the study documented that cigarette smoking reduced Cmax and increased T1/2 values [17]. Specific genotypes of cytochrome P450 (CYP) enzymes also altered pharmacokinetics [17].
The effect of cigarette smoking was also investigated in a cohort study including eight habitual smokers [19]. The volunteers received 25 mg of oral melatonin daily during periods of smoking and smoking-free periods, respectively. Cigarette smoking reduced Cmax from 1858 pg/ml (smoking-free period) compared to 640 pg/ml (smoking period). Also, AUC was reduced from 294,002 pg/ml × min during smoking-free periods to 102,419 pg/ml × min during periods of smoking. Tmax values were identical between the groups [19].
The effect of oral contraceptives (OC) and genotype of CYP-enzymes was investigated in a cohort study including 29 volunteers [23]. Following oral administration of 6 mg of melatonin, OC significantly increased Cmax and AUC and reduced Cl/F values [23]. Furthermore, the genotype of CYP-enzymes (wild-type allele or variant allele) was assessed. Genotypes did not affect the pharmacokinetics of melatonin [23].
A cohort study including 12 volunteers administered 2 mg of oral melatonin in different oral formulations in fed and fasting state, respectively [25]. Cmax and AUC were increased during fed state; however, statistical comparisons were not performed. The study documented similar Tmax values between the groups. T1/2 ranged from 32 (gelatin-coated capsules, fasting state) to 40 min (corn-oil preparation, fed state).
The effect of co-administration of 50 mg of fluvoxamine and 5 mg of melatonin was investigated in a cohort study including five male volunteers [29]. Cmax, AUC, and T1/2 values were significantly increased with the co-administration of fluvoxamine.
Pharmacokinetics in patients
The pharmacokinetics of melatonin has been investigated in three studies investigating critically ill patients [3, 16] and elderly patients with insomnia [21], respectively.
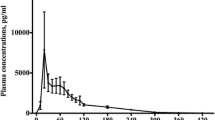
A RCT investigating 24 critically ill patients administered either 10 mg of oral melatonin or placebo [3]. The melatonin group exhibited a Cmax of 14,974 pg/ml, a Tmax of 30 min, a T1/2 of 88 min, and an AUC of 1.80 × 106 pg/ml × min.
Mistraletti and colleagues investigated a cohort of 12 critically ill patients [16]. An oral dose of 3 mg melatonin was administered as crushed tablets by a nasogastric tube. The study demonstrated a Cmax of 11,040 pg/ml, a Tmax of 16 min, a T1/2 of 94 min, and an AUC of 1.69 × 106 pg/ml × min.
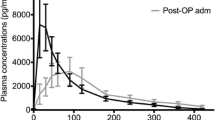
A two-arm RCT investigated 27 elderly patients suffering from insomnia [21]. The study administered two surge-sustained release formulations (dose 0.4/4 mg), respectively. Cmax and AUC were significantly higher in the high-dose group compared to the low-dose group. No differences in Tmax, T1/2, Cl/F, or VD/F values were demonstrated between the groups.
Discussion
The pharmacokinetics of exogenous melatonin has primarily been investigated in healthy volunteers. The studies investigating oral immediate-release formulations demonstrated Tmax values of approximately 50 min. A T1/2 of approximately 45 min was documented following both oral and intravenous administrations. Studies administering oral slow-release formulations demonstrated prolonged Tmax and T1/2 values of up to 167 and 91 min, respectively.
Our review displayed extensive variations within- and between studies with respect to the pharmacokinetic variables, Cmax, AUC, Cl, and VD. These variations may obviously be caused by individual differences in absorption, distribution, metabolism, and elimination of the drug, though further details are not known at this moment [9, 20]. It should, however, be investigated, if these variations may potentially affect drug efficacy. Interestingly, no link between specific melatonin plasma concentration levels and actual clinical effects (or adverse effects) has been established yet. Furthermore, the low number of included studies and the differences in study designs, dosages (0.3–100 mg), administration routes (oral and IV), drug formulations, and melatonin analysis assays may also have contributed to the variability of data. Regarding the use of different analysis assays, it might be a significant factor to the observed variability of the results. Interestingly, no studies performing direct comparisons of the different assays have performed so far.
Our review also demonstrated a low bioavailability [8, 9]. It may either result from low absorption from the gastrointestinal canal, from an extensive first-pass metabolism in the liver or from a combination of both [9]. At this moment, the exact intestinal absorption fraction of oral melatonin in humans is not established [20]. On the other hand, Di and colleagues documented an increased metabolite production following oral administration compared to intravenous, indicating first-pass metabolism as the primary determinant [9]. Assessments of bioavailability may, however, also be affected by the administration of different drug formulations. It is crucial that the measuring periods should be adapted to the prolonged absorption phase of e.g., slow-release formulations in order not to underestimate the bioavailability of the drug (or at least estimate bioavailability by applying the elimination constant and last measured plasma/serum concentration). Future studies investigating bioavailability should attempt to provide detailed pharmacokinetic analyses of metabolites following melatonin administration by different administration routes. Moreover, further investigations of the physiological and possible clinical effects of melatonin metabolites, such as 6-sulfatoxymelatonin, N-acetyl-5-methoxykynuramine (AMK), and N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) are needed [30]. Conclusively, studies also indicated a substantial intra-study variability of bioavailability, demonstrated by values ranging between 10 and 56 % [9]. This variability was, however, not present to the same extend in a recent high-quality study [8]. Future studies should examine, if inter-individual variations of bioavailability are actually present, or caused by methodological differences between the studies.
Our review also documented that external factors, such as caffeine [17], cigarette smoking [19], OC [23] and fluvoxamine [29] either increased [17, 23, 29] or reduced Cmax and AUC values of melatonin. Correspondingly, elimination rates (T1/2 and Cl) were reduced [23, 29] or increased [17]. In this case, the altered pharmacokinetics probably results from an altered metabolism by specific CYP-enzymes in the liver [17, 19, 29]. Especially, the CYP 1A2 enzyme seems to play a key role in the metabolism of melatonin. Other external factors, such as fasting also reduced Cmax compared to fed state [25]. These findings may influence future dosing regimens in surgical patients receiving perioperative melatonin treatments [2, 25]. In critically ill patients, an accelerated Tmax and extended T1/2 were demonstrated, probably due to a reduced liver and kidney function in this patient category [3, 16]. In general, the effects of external factors may mandate alternative dosing regimens in selected patient groups. Otherwise, risks of inadequate drug plasma concentrations or clinical “hangover” effects due to increased/prolonged plasma concentrations may occur. It should, however, be underlined that studies in “standard” medical (without serious comorbidity) and surgical patients should be performed before differences in pharmacokinetics between volunteers and patients can be finally settled.
This review was performed in accordance to PRISMA guidelines [10]. It is the first systematic review to provide a comprehensive overview of the literature concerning the pharmacokinetics of melatonin in adult volunteers and patients. We chose not to include a quality or risk of bias analysis due to the heterogeneous nature of the included studies (RCTs, cohort and case reports studies in both volunteers and patient) [31, 32]. Furthermore, the pharmacokinetic variables assessed in this review do not relate to potential bias or quality parameters (e.g., randomization and blinding) evaluated by these scoring systems. Finally, no quantitative analysis was feasible, also due to the extensive variation in study designs, analysis methods, dosages, and administration routes.
In conclusion, this review documented a Tmax of approximately 50 min following oral immediate-release formulations of melatonin. T1/2 of both oral and intravenous melatonin was about 45 min. An administration of melatonin 45 min before intended clinical effect may therefore be advocated. A clinician should also consider external factors, which may potentially affect the pharmacokinetics of melatonin. Cmax, AUC, Cl, and VD displayed extensive variation within and between studies. The variations may obviously relate to inter-individual differences in drug absorption, distribution, metabolism, and elimination but may also be confounded by substantial variability in study designs/methods. Bioavailability was generally low (approximately 15 %) and potentially with a significant intra-individual variability.
References
Brzezinski A (1997) Melatonin in humans. N Engl J Med 336:186–195
Andersen LPH, Werner MU, Rosenberg J, Gögenur I (2014) A systematic review of peri-operative melatonin. Anaesthesia 69:1163–1171
Bourne RS, Mills GH, Minelli C (2008) Melatonin therapy to improve nocturnal sleep in critically ill patients: encouraging results from a small randomized controlled trail. Crit Care 12:R52
Borazan H, Tuncer S, Yalcin N, Erol A, Otelcioglu S (2010) Effects of preoperative oral melatonin medication on postoperative analgesia, sleep quality, and sedation in patients undergoing elective prostatectomy: a randomized clinical trial. J Anesth 24:155–160
Caumo W, Torres F, Moreia NL, Auzani JAS, Monteiro CA, Londero G, Ribeiro DFM, Hidalgo MPL (2007) The clinical impact of preoperative melatonin on postoperative outcomes in patients undergoing abdominal hysterectomy. Anesth Analg 105:1263–1271
Gitto E, Romeo C, Reiter RJ, Impellizzeri P, Pesce S, Basile M, Antonuccio P, Trimarchi G, Gentile C, Barberi I, Zuccarello B (2004) Melatonin reduces oxidative stress in surgical neonates. J Pediatr Surg 39:184–189
Kücükakin B, Lykkesfeldt J, Nielsen HJ, Reiter RJ, Rosenberg J, Gögenur I (2008) Utility of melatonin to treat surgical stress after major vascular surgery—a safety study. J Pineal Res 44:426–431
DeMuro RL, Nafziger AN, Blask DE, Menhinick AM, Bertino JS (2000) The absolute bioavailability of oral melatonin. J Clin Pharmacol 40:781–784
Di WL, Kadva A, Johnston A, Silman R (1997) Variable bioavailability of oral melatonin. N Engl J Med 336:1028–1029
Moher D, Liberati A, Tetzlaff J, Altman DG (2010) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Int J Surg 8:336–341
Zhdanova IV, Wurtman RJ, Balcioglu A, Kartashov AI, Lynch HJ (1998) Endogenous melatonin levels and the fate of exogenous melatonin: age effects. J Gerontol 53:B293–B298
Cavallo A, Ritschel WA (1996) Pharmacokinetics of melatonin in human sexual maturation. J Clin Endocrinol Metab 81:1882–1886
Vakkuri O, Leppäluoto J, Kauppila A (1985) Oral administration and distribution of melatonin in human serum, saliva and urine. Life Sci 37:489–495
Shirakawa SI, Tsuchiya S, Tsutsumi Y, Kotorii T, Uchimura N, Sakamoto T, Yamada S (1998) Time course of saliva and serum melatonin levels after ingestion of melatonin. Psychiatry Clin Neurosci 52:266–267
Waldhauser F, Waldhauser M, Lieberman HR, Deng MH, Lynch HJ, Wurtman RJ (1984) Bioavailability of oral melatonin in humans. Neuroendocrinology 39:307–311
Mistraletti G, Sabbatini G, Taverna M, Figini MA, Umbrello M, Magni P, Ruscica M, Dozio E, Esposti R, DeMartini G, Fraschini F, Rezzani R, Reiter RJ, Iapichino G (2010) Pharmacokinetics of orally administered melatonin in critically ill patients. J Pineal Res 48:142–147
Härtter S, Nordmark A, Rose DM, Bertilsson L, Tybring G, Laine K (2003) Effects of caffeine intake on the pharmacokinetics of melatonin, a probe drug for CYP1A2 activity. Br J Clin Pharmacol 56:679–682
Mallo C, Zaîdan R, Galy G, Vermeulen E, Brun J, Chazot G, Claustrat B (1990) Pharmacokinetics of melatonin in man after intravenous infusion and bolus injection. Eur J Clin Pharmacol 38:297–301
Ursing C, Bhar CV, Brismar K, Röjdmark S (2005) Influence of cigarette smoking on melatonin levels in man. Eur J Clin Pharmacol 61:197–201
Fourtillan JB, Brisson AM, Gobin P, Ingrand I, Decourt JPH, Girault J (2000) Bioavailability of melatonin in humans after day-time administration of D7 Melatonin. Biopharm Drug Dispos 21:15–22
Gooneratne NS, Edwards AYZ, Zhou C, Cuellar N, Grandner MA, Barrett JS (2012) Melatonin pharmacokinetics following two different oral surge-sustained release doses in older adults. J Pineal Res 52:437–445
Markantonis SL, Tsakalozou E, Paraskeva A, Staikou C, Fassoulaki A (2008) Melatonin pharmacokinetics in premenopausal and postmenopausal healthy female volunteers. J Clin Pharmacol 48:240–245
Hilli J, Korhonen T, Turpeinen M, Hokkanen J, Mattila S, Laine K (2008) The effect of oral contraceptives on the pharmacokinetics of melatonin in healthy subjects with CYP1A2 g.-163C > A polymorphism. J Clin Pharmacol 48:986–994
Le Bars D, Thivolle P, Vitte PA, Bojkowski C, Chazot G, Arendt J, Frackowiak RSJ, Claustrat B (1991) PET and plasma pharmacokinetic studies after bolus intravenous administration of [11C]melatonin in humans. Nucl Med Biol 18:357–362
Aldhous M, Franey C, Wright J, Arendt J (1985) Plasma concentrations of melatonin in man following oral absorption of different preparations. J Clin Pharmacol 19:517–521
López-Gamboa M, Canales-Gómez JS, Castro Sandoval TJ, Tovar EN, Mejía MA, Baltazar MAM, Palma-Aguirre JA (2010) Bioavailability of long acting capsules of melatonin in Mexican healthy volunteers. J Bioequiv Availab 2:116–119
Proietti S, Carlomagno G, Dinicola S, Bizzarri M (2014) Soft gel capsules improve melatonin’s bioavailability in humans. Expert Opin Drug Metab Toxicol 10:1193–1198
Hoffmann H, Dittgen M, Hoffmann A, Bartsch C, Breitbarth H, Timpe C, Farker K, Schmidt U, Mellinger U, Zimmermann H, Gräser T, Oettel M (1998) Evaluation of an oral pulsatile delivery system for melatonin in humans. Pharmazie 53:462–466
Härrter S, Grözinger M, Wiegmann H, Röschke J, Hiemke C (2000) Increased bioavailability of oral melatonin after fluvoxamine coadministration. Clin Pharmacol Ther 67:1–6
Tan DX, Manchester LC, Terron MP, Flores LJ, Reiter RJ (2007) One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res 42:28–42
Higgins JPT, Green S (eds) (2011) Cochrane handbook for systematic reviews of interventions version 5.1.0 [updated March 2011]. The Cochrane Collaboration. http://www.cochrane-handbook.org. Accessed 16 Dec 2014
Jadad AR, Moore RA, Carrol D, Jenkinson C, Reynolds JM, Gavaghan DJ, McQuay HJ (1996) Assessing the quality of reports of randomized clinical trails: is blinding necessary? Contemp Clin Trails 17:1–12
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Harpsøe, N.G., Andersen, L.P.H., Gögenur, I. et al. Clinical pharmacokinetics of melatonin: a systematic review. Eur J Clin Pharmacol 71, 901–909 (2015). https://doi.org/10.1007/s00228-015-1873-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-015-1873-4