
Abstract
Despite widespread clinical application of melatonin, several unanswered questions remain regarding the pharmacokinetics of this drug. This lack of knowledge may contribute to the inconsistency of results in previous clinical studies. Currently, a t max value of 30–45 min and a t ½elimination of 45 min are well established. Several questions relate to what constitutes a clinically effective plasma concentration, the choice of ideal administration route, and the optimal method of analysis. Furthermore, investigations of melatonin metabolites in humans are urgently needed in order to characterize their biological functions and the metabolic fates of these derivatives. Finally, pharmacokinetics in patients should be investigated further in order to reduce the risk of potential adverse effects, such as daytime sleepiness or unintended sedation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
A t max of 30–45 min of oral melatonin is well established. |
The t ½elimination of melatonin is 45 min. |
The pharmacokinetics of melatonin in patients needs to be investigated further. |
1 Introduction
Exogenous melatonin has been administered for various diseases, such as insomnia [1], hypertension [2], metabolic syndrome [3], chronic obstructive pulmonary disease [4], and inflammatory bowel disease [5]. Its use in surgical patients has also demonstrated anxiolytic, analgesic and antioxidative properties of the drug [6–9]. In addition, a number of studies indicate possible regulation of sleep and delirium in critically ill patients [10].
Unexpectedly, the findings in clinical studies have been conflicting [6], despite an abundance of evidence from experimental research implying that it could be an effective treatment [11, 12]. Interestingly, recent studies indicate that genetic variants coding melatonin receptors may, in part, cause these differences [13]. Currently, exogenous melatonin is therefore only recommended routinely as a sleeping aid in primary and secondary sleep disorders [14]. Clearly, a number of unanswered questions concerning possible future indications of melatonin and its mechanisms of action remain [15]. A fundamental step towards clinical efficacy of any drug is a thorough understanding of its pharmacokinetic properties.
2 Could Variable Pharmacokinetics Explain The Discrepancy In Clinical Effect?
Only a limited number of studies have been performed investigating the pharmacokinetic properties in humans [16]. These studies primarily included young healthy subjects who were administered various doses and formulations, employed different pharmacokinetic analyses, and reported a variety of pharmacokinetic variables [16]. Harpsøe and colleagues have previously reviewed their findings in detail [16], although a number of imperative questions relating to pharmacokinetic properties and clinical efficacy still remain. This paper aims to address these scientific questions in order to augment future clinical melatonin research.
Despite the major differences in study designs, time to reach maximal plasma concentrations (t max) and elimination half-life (t ½elimination) of melatonin in humans are well established [16]. Oral melatonin is rapidly absorbed from the small intestine by first-order kinetics, and t max is achieved after approximately 30–45 min [16]. An oral administration 45 min before intended effects may therefore still be advocated if maximal plasma concentrations (C max) coincide with maximal clinical effects; however, this correlation needs to be established in future studies.
Similarly, a t ½elimination of approximately 45 min has been documented in several studies in a wide range of doses, up to 100 mg intravenously [16]. This parameter may also be described by first-order elimination kinetics, and is independent of dose and route of administration [16]. Interestingly, an experimental study in prepubertal subjects suggested an accelerated elimination in children compared with adults [17]. Another clinical study in critically ill patients documented an impeded elimination, probably owing to altered liver and kidney function in this patient group [18]. The clinical impact of these findings in different subject/patient groups remains to be established.
A generally low bioavailability (f) of oral melatonin has been documented in a number of studies, although displaying variable mean values ranging from 3 to 33 % [16]. Similarly, studies demonstrated a substantial intrastudy variation between subjects [19]. A recent experimental report recorded values ranging between 0.7 and 12.7 % [20]. It is generally agreed that the low f is caused by a considerable first-pass metabolism in the liver [19]. Di and colleagues addressed this issue by measuring the production of the main metabolite 6-hydroxymelatonin sulphate following oral and intravenous administration [19]. Compared with intravenous injection, oral administration provided an increased metabolite/melatonin plasma level ratio, indicating liver metabolism as the main determinant of the low oral f [19]. However, the exact values and applied methods were not reported in this small study. The significance of liver metabolism was also emphasized by other reports in which coadministration of cytochrome P450 (CYP) 1A2 enzyme drug substrates, such as fluvoxamine [21], caffeine [22], or oral contraceptives [23], along with exogenous melatonin, had a substantial impact on subsequent plasma melatonin levels. Finally, studies in liver cirrhosis patients also demonstrated reduced elimination rates and increased plasma melatonin levels [24].
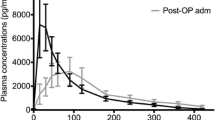
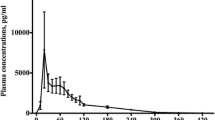
In general, the reported C max and area under the curve from time zero to infinity (AUC∞) values also varied extensively between studies as a result of differences in experimental designs [16]. Also of note, these values were highly variable between subjects following identical dosing regimens. For example, following 10 mg of oral melatonin, maximal plasma levels ranged between 1105 and 58,900 pg/ml [20]. The exact cause of this extreme variability is unknown but may obviously relate to interindividual differences in absorption, distribution, metabolism, and elimination of the drug. Correspondingly, clearance (CL) and volume of distribution (V d) following intravenous administrations fluctuated extensively but may, at this moment, be estimated to be approximately 0.025 L min−1 kg−1 and 2.0 L kg−1, respectively [16, 20].
3 Unanswered Questions
Several unanswered questions remain with respect to analytical methods, clinically relevant plasma levels, optimal administration routes, pharmacokinetics and function of melatonin metabolites, and, finally, melatonin administration in patient groups.
First, the studies investigating plasma melatonin levels have applied various methods of analysis, including radioimmunoassay, enzyme-linked immunosorbent assay, and mass spectrometry [16]. Unfortunately, no studies have performed a direct comparison of the individual methods assessing their construct validity in humans.
Second, as mentioned above, plasma melatonin levels varied extensively within studies, even with identical doses. These substantial differences may potentially impact clinical efficacy, with only one clinical study to date correlating clinical effects and actual plasma melatonin concentrations following exogenous administration [25]. In this context, it should be underlined that dosage, subsequent plasma concentrations, and possible clinical effects may very well vary for different indications. As an example, clinical studies demonstrated the antioxidative/anti-inflammatory effects of melatonin following administration of multiple large doses (10 mg/kg × 10 intravenously) [8] compared with the low single doses applied in sleep-aid dosing regimens (approximately 1–10 mg orally) [1]. Therefore, in-depth pharmacokinetic/pharmacodynamic modeling should be performed within the individual indications. Similarly, genetic variation and receptor function has been shown to affect clinical effects in experimental setups [13], although the possible clinical impact in patients is still unknown. However, interestingly, reduced levels of endogenous melatonin have been established in various conditions, such as ischemic heart disease, advanced diabetes, and certain types of cancers [26–28]. Nonetheless, these findings originate from cohort studies, and the possible implications are therefore limited by the unestablished causality.
Third, these data could also provide needed answers with regard to optimal administration routes for melatonin. At this moment, pharmacokinetic studies have only investigated oral and intravenous administrations [16]. Alternative routes, such as sublingual, intranasal, or rectal administration may be ideal in a clinical setting by encompassing a rapid t max, a high f, and an uncomplicated administration procedure.
Fourth, to date, studies have assessed either direct plasma melatonin concentrations or urinary levels of the main metabolite, 6-hydroxymelatonin sulphate. Only two preliminary small studies have provided a rough estimate of this metabolite in the plasma of humans [19, 23]. A thorough investigation of this and other metabolites is required in order to further characterize metabolism and elimination of melatonin following its administration, preferably comparing different administration routes. Even more importantly, recent studies provide evidence that melatonin metabolites are physiologically- and possibly also clinically-active compounds [29].
Fifth, only four clinical studies have estimated melatonin pharmacokinetics in critically ill patients [18, 30, 31] and elderly suffering from insomnia [32]. The studies demonstrated that elimination in frail patients was impeded, and a potential risk of extended supraphysiological melatonin plasma levels and potential overdose issues were present. Although no serious adverse effects have been documented from melatonin treatment in patients, the potential risks of, for example, day-time sleepiness and/or unintended sedation should be addressed by dose regulation. These findings may obviously mandate further investigations in other patient groups in order to tailor dosing regimens to specific patient groups and indications.
As a final consideration, now that melatonin is known to be present in edible plants and commercially available meat products [33, 34], consideration should be given to standardizing the diet of subjects being tested for the pharmacokinetic properties when melatonin is administered. Similarly, future studies should also identify patients receiving either fluvoxamine [21] or oral contraceptives [23], and potentially also regulate caffeine intake [22] and smoking [35], which may otherwise introduce bias between investigated subjects.
4 Conclusions
Despite widespread clinical application of melatonin, several unanswered questions remain regarding the pharmacokinetics of this drug. This lack of knowledge may contribute to the inconsistency of results in previous clinical studies. Currently, a t max value of 30–45 min and a t ½elimination of 45 min are well established. However, several questions relate to what constitutes a clinically effective plasma concentration, the choice of ideal administration route, and the optimal method of analysis. Furthermore, investigations of melatonin metabolites in humans are urgently needed in order to characterize their biological functions and the metabolic fates of these derivatives. Finally, pharmacokinetics in patients should also be investigated further in order to reduce the risk of potential adverse effects, such as daytime sleepiness or unintended sedation.
References
Brzezinski A, Vangel MG, Wurtman RJ, Norrie G, Zhdanova I, Ben-Shushan A, et al. Effects of exogenous melatonin on sleep: a meta-analysis. Sleep Med Rev. 2005;9:41–50.
Grossman E, Laudon M, Yalcin R, Zengil H, Peleg E, Sharabi Y, et al. Melatonin reduces night blood pressure in patients with nocturnal hypertension. Am J Med. 2006;119:898–902.
Koziróg M, Poliwczak AR, Duchnowicz P, Koter-Michalak M, Sikora J, Broncel M. Melatonin treatment improves blood pressure, lipid profile, and parameters of oxidative stress in patients with metabolic syndrome. J Pineal Res. 2011;50:261–6.
De Matos Cavalcante AG, de Bruin PF, de Bruin VM, Nunes DM, Pereira ED, Cavalcante MM, et al. Melatonin reduces lung oxidative stress in patients with chronic obstructive pulmonary disease: a randomized, double-blind, placebo-controlled study. J Pineal Res. 2012;53:238–44.
Song GH, Leng PH, Gwee KA, Moochhala SM, Ho KY. Melatonin improves abdominal pain in irritable bowel syndrome patients who have sleep disturbances: a randomised, double blind, placebo controlled study. Gut. 2005;54:1402–7.
Andersen LPH, Werner MU, Rosenberg J, Gögenur I. A systematic review of peri-operative melatonin. Anaesthesia. 2014;69:1163–71.
Gögenur I, Kücükakin B, Panduro Jensen L, Reiter RJ, Rosenberg J. Melatonin reduces cardiac morbidity and markers of myocardial ischemia after elective abdominal aortic aneurism repair: a randomized, placebo-controlled, clinical trial. J Pineal Res. 2014;57:10–5.
Gitto E, Romeo C, Reiter RJ, Impellizzeri P, Pesce S, Basile M, et al. Melatonin reduces oxidative stress in surgical neonates. J Pediatr Surg. 2004;39:184–9.
Gitto E, Karbownik M, Reiter RJ, Tan DX, Cuzzocrea S, Chiurazzi P, et al. Effects of melatonin treatment in septic newborns. Pediatr Res. 2001;50:756–60.
Bellapart J, Boots R. Potential use of melatonin in sleep and delirium in the critically ill. Br J Anaesth. 2012;108:572–80.
Srinivasan V, Pandi-Perumal SR, Trahkt I, Spence DW, Poeggeler B, Hardeland R, et al. Melatonin and melatonergic drugs on sleep: possible mechanisms of action. Int J Neurosci. 2009;119:821–46.
Srinivasan V, Pandi-Perumal SR, Spence DW, Kato H, Cardinali DP. Melatonin in septic shock: some recent concepts. J Crit Care. 2010;25(656):e1–6.
Garaulet M, Gómez-Abellán P, Rubio-Sastre P, Madrid JA, Saxena R, Scheer FA. Common type 2 diabetes risk variant in MTNR1B worsens the deleterious effect of melatonin on glucose tolerance in humans. Metabolism. 2015;64:1650–7.
Arendt J, Skene DJ. Melatonin as a chronobiotic. Sleep Med Rev. 2005;9:25–39.
Andersen LPH, Rosenberg J, Gögenur I. Perioperative melatonin: not ready for prime time. Br J Anaesth. 2014;112:7–8.
Harpsøe NG, Andersen LPH, Gögenur I, Rosenberg J. Clinical pharmacokinetics of melatonin: a systematic review. Eur J Clin Pharmacol. 2015;71:901–9.
Cavallo A, Ritschel WA. Pharmacokinetics of melatonin in human sexual maturation. J Clin Endocrinol Metab. 1996;81:1882–6.
Bourne RS, Mills GH, Minelli C. Melatonin therapy to improve nocturnal sleep in critically ill patients: encouraging results from a small randomised controlled trial. Crit Care. 2008;12:R52.
Di WL, Kadva A, Johnston A, Silman R. Variable bioavailability of oral melatonin. N Engl J Med. 1997;336:1028–9.
Andersen LP, Werner MU, Rosenkilde MM, Harpsøe NG, Fuglsang H, Rosenberg J, et al. Pharmacokinetics of oral and intravenous melatonin in healthy volunteers. BMC Pharmacol Toxicol. 2016;17:8.
Härtter S, Grözinger M, Weigmann H, Röschke J, Hiemke C. Increased bioavailability of oral melatonin after fluvoxamine coadministration. Clin Pharmacol Ther. 2000;67:1–6.
Härtter S, Nordmark A, Rose DM, Bertilsson L, Tybring G, Laine K. Effects of caffeine intake on the pharmacokinetics of melatonin, a probe drug for CYP1A2 activity. Br J Clin Pharmacol. 2003;56:679–82.
Hilli J, Korhonen T, Turpeinen M, Hokkanen J, Mattila S, Laine K. The effect of oral contraceptives on the pharmacokinetics of melatonin in healthy subjects with CYP1A2 g.-163C > A polymorphism. J Clin Pharmacol. 2008;48:986–94.
Lane EA, Moss HB. Pharmacokinetics of melatonin in man: first pass hepatic metabolism. J Clin Endocrinol Metab. 1985;61:1214–6.
Stefani LC, Muller S, Torres IL, Razzolini B, Rozisky JR, Fregni F, et al. A phase II, randomized, double-blind, placebo controlled, dose-response trial of the melatonin effect on the pain threshold of healthy subjects. PLoS One. 2013;8:e74107.
Yaprak M, Altun A, Vardar A, Aktoz M, Ciftci S, Ozbay G. Decreased nocturnal synthesis of melatonin in patients with coronary artery disease. Int J Cardiol. 2003;89:103–7.
Hikichi T, Tateda N, Miura T. Alteration of melatonin secretion in patients with type 2 diabetes and proliferative diabetic retinopathy. Clin Ophthalmol. 2011;5:655–60.
Schernhammer ES, Hankinsin SE. Urinary melatonin levels postmenopausal breast cancer risk in the nurses’ health study cohort. Cancer Epidemiol Biomark Prev. 2009;18:74–9.
Galano A, Tan DX, Reiter RJ. On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J Pineal Res. 2013;54:245–57.
Mistraletti G, Sabbatini G, Taverna M, Figini MA, Umbrello M, Magni P, et al. Pharmacokinetics of orally administered melatonin in critically ill patients. J Pineal Res. 2010;48:142–7.
Bellapart J, Roberts JA, Appadurai V, Wallis SC, Nuñez-Nuñez M, Boots RJ. Pharmacokinetics of a novel dosing regimen of oral melatonin in critically ill patients. Clin Chem Lab Med. 2016;54:467–72.
Gooneratne NS, Edwards AYZ, Zhou C, Cuellar N, Grandner MA, Barrett JS. Melatonin pharmacokinetics following two different oral surge-sustained release doses in older adults. J Pineal Res. 2012;52:437–45.
Reiter R, Tan DX, Zhou Z, Cruz M, Fuentes-Broto L, Galano A. Phytomelatonin: assisting plants to survive and thrive. Molecules. 2015;20:7396–437.
Tan DX, Zanghi BM, Manchester LC, Reiter RJ. Melatonin identified in meats and other food stuffs: potentially nutritional impact. J Pineal Res. 2014;57:213–8.
Ursing C, von Bahr C, Brismar K, Röjdmark S. Influence of cigarette smoking on melatonin levels in man. Eur J Clin Pharmacol. 2005;61:197–201.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflicts of interest
Lars P. H. Andersen, Ismail Gögenur, Jacob Rosenberg and Russel J. Reiter declare that they have no conflicts of interest that might be relevant to the contents of this article.
Rights and permissions
About this article

Cite this article
Andersen, L.P.H., Gögenur, I., Rosenberg, J. et al. Pharmacokinetics of Melatonin: The Missing Link in Clinical Efficacy?. Clin Pharmacokinet 55, 1027–1030 (2016). https://doi.org/10.1007/s40262-016-0386-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0386-3