
Abstract
Background
The extent of P2Y12 inhibition during coronary intervention is an important determinant of ischemic complications. The currently available oral P2Y12 inhibitors are limited by a relatively slow onset of action and variable on-treatment response.
Objective
Our objective was to determine the pharmacodynamic (PD) dose–antiplatelet response relationship and the pharmacokinetics of MDCO-157, an intravenous formulation of clopidogrel complexed with sulphobutylether betacyclodextrin, and to identify the dose level of MDCO-157 that matches the PD effect of oral clopidogrel 300 mg.
Methodology
A randomized open-label crossover study was performed in 33 healthy adult volunteers to determine the pharmacokinetic (clopidogrel and clopidogrel H4 thiol active metabolite) and the PD (vasodilator-stimulated phosphoprotein [VASP]) effects of MDCO-157 at doses of 75, 150, and 300 mg and of oral clopidogrel 300 mg.
Results
Data are presented as %, mean (standard deviation). The maximum effect of P2Y12 receptor inhibition assessed by flow cytometry using VASP was 70.42 (6.7), 69.45 (7.1), and 65.58 (12.6) for intravenous MDCO-157 at doses of 75, 150, and 300 mg, respectively, compared with 56.6 (17.5) with oral clopidogrel 300 mg administration (p < 0.0001). Intravenous administration of MDCO-157 led to a stepwise increase in plasma exposure of clopidogrel, higher than with administration of an oral dose of 300 mg (p < 0.0001). Plasma exposure of H4-thiol also increased with intravenous dose (3.6 ± 2.6, 6.9 ± 4.6, and 12.4 ± 9.1 h·ng/ml for intravenous 75, 150, and 300 mg, respectively) but was lower than with oral administration of a 300-mg dose (34.0 ± 16.0 h.ng/ml; pairwise p < 0.0001).
Conclusions
MDCO-157, an intravenous formulation of clopidogrel complexed with sulphobutylether betacyclodextrin, did not show significant platelet inhibition when administered at doses up to 300 mg. Higher doses with longer infusion may be needed to reach a sufficient threshold of active metabolite generation.
Trial Registration: ClinicalTrials.gov identifier: NCT01860105.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
MDCO-157, an intravenous formulation of clopidogrel bisulfate, produce pharmacodynamic transient and minor effects that were much lower in magnitude than that of oral clopidogrel 300 mg. |
This was due to an insufficient threshold of active metabolite generation as a consequence of the extrahepatic clearance of the drug. |
Whether a higher dose would overcome this limitation and possibly be clinically relevant is unknown. |
1 Introduction
Clopidogrel is a second-generation thienopyridine that irreversibly inhibits the purinergic P2Y12 adenosine diphosphate (ADP) platelet membrane receptor [1, 2]. It is an inactive pro-drug that requires oxidation by the hepatic cytochrome P450 (CYP) system to generate clopidogrel H4-thiol, the putative only active metabolite [3]. It has been the standard of care in combination with aspirin to prevent cardiovascular events in patients with an acute coronary syndrome (ACS) and/or undergoing percutaneous coronary intervention (PCI) and remains the most extensively used P2Y12 receptor inhibitor [4]. Although more potent P2Y12 inhibitors with a faster onset, namely prasugrel and ticagrelor, demonstrated superiority over clopidogrel to reduce recurrent ischemic events, these oral formulations are associated with limitations due to delayed absorption, particularly in STEMI patients when morphine is concomitantly used, and more bleeding events [5–10]. Intravenous agents with faster onset of action have been developed to overcome these limitations in the acute care setting [11]. Cangrelor, an intravenous adenosine triphosphate (ATP) analog that binds reversibly and with high affinity to the platelet P2Y12 receptor and has a short plasma half-life (<10 min), is now approved for patients who have not received an oral P2Y12 inhibitor prior to the PCI procedure and in whom oral therapy with P2Y12 inhibitors is not feasible or desirable [12–14]. However, there remains biological uncertainty with regards to the transition from cangrelor to oral P2Y12 inhibitor [15–18].
MDCO-157 is an intravenous formulation of clopidogrel complexed with an excipient, sulphobutylether betacyclodextrin (Captisol® Ligand, La Jolla, CA, USA), which increases the aqueous solubility and stability of clopidogrel. Sulphobutylether betacyclodextrin is used in four marketed parenteral products. MDCO-157 was developed with the intention of matching the antiplatelet effects of oral clopidogrel. A previous clinical study demonstrated that MDCO-157 produced a rapid onset of platelet aggregation inhibition at doses greater than 30 mg, which was sustained through the 24-h test period [19]. With this intravenous formulation, dose-proportional increases were seen in the exposure parameters for clopidogrel, the carboxylic acid metabolite (inactive metabolite), and the clopidogrel H4-thiol metabolite. MDCO-157 was well tolerated at all doses. However, methodology limitations led to inconclusive pharmacodynamic (PD) results.
The AMPHORE (Appraisal of MDCO-157 and clopidogrel pharmacokinetics and pharmacodynamics in Healthy volunteers with an Open-label, Randomized, cross-over Evaluation) study was thus designed as a four-period, cross-over PD and pharmacokinetic (PK) study in healthy volunteers using multiple assay methodologies for measuring platelet aggregation and P2Y12 inhibition. Clopidogrel 300 mg as well as MDCO-157 doses of 75, 150, and 300 mg of MDCO-157 were evaluated. AMPHORE was built upon previous clinical experience with MDCO-157 and clopidogrel while incorporating multiple within-study controls and self-controls between subjects to enhance the reliability and analysis of data. The primary objectives were to determine the dose-response in the PD effect of MDCO 157 and to identify the dose level of MDCO-157 that matches the PD effect of clopidogrel 300 mg. The secondary objectives were to determine the PK of MDCO-157, including clopidogrel, clopidogrel carboxylic acid, and clopidogrel H4-thiol active metabolite in plasma and to assess the safety and tolerability of single doses of MDCO 157.
2 Methods
2.1 Study Design
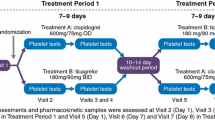
The AMPHORE study was a phase I, open-label, randomized, four-period cross-over study to determine the PD and PK of MDCO-157 and clopidogrel in adult healthy volunteers. Intravenous MDCO-157 doses of 75, 150, and 300 mg were infused over 2.5–10 min and a clopidogrel 300 mg tablet was administered to each subject in a randomized Williams design [20] with four sequences (Fig. 1). Each study period was separated by a washout period of 14 ± 2 days.

Design of AMPHORE 4 × 4 cross-over study
2.2 Study Endpoints
The primary endpoints of this trial were maximum effect of P2Y12 receptor inhibition (E max) using vasodilator-stimulated phosphoprotein (VASP) (flow cytometry) and area under the effect of P2Y12 receptor inhibition time curve (AUEC) using VASP (flow cytometry). The secondary endpoints were maximum effect of inhibition of platelet aggregation using light transmission aggregometry (LTA), area under the effect of inhibition of platelet aggregation time curve using LTA, maximum effect of inhibition of platelet aggregation using VerifyNow P2Y12 (VN-P2Y12, Accumetrics, San Diego, CA, USA), and area under the effect of inhibition of platelet aggregation time curve using VN-P2Y12.
2.3 Selection of Healthy Volunteers
To enter the AMPHORE study, healthy male and female individuals aged 18–45 years were screened at the Clinical Investigation Center Paris-Est (CIC-1421), the Institut de Cardiologie of Pitié-Salpétrière Hospital, and the Medical School of the Pierre et Marie Curie University. Major exclusion criteria were smoking habits; any history of bleeding or coagulation disorders; use of aspirin, non-steroidal anti-inflammatory drugs, CYP3A4 inhibitors (e.g., ketoconazole), CYP2C19 inhibitors (e.g., omeprazole), or other drugs known to affect platelet function or coagulation within 14 days prior to receiving THE study drug (MDCO-157 or clopidogrel). Women of childbearing potential had to be using a medically acceptable form of birth control throughout the study, and the absence of pregnancy was verified at each study period. Other inclusion criteria included a body mass index between 18 and 35 kg/m2, the absence of significant disease or abnormal standard laboratory values, and a normal 12-lead electrocardiogram (ECG). Finally, other important exclusion criteria included known hypersensitivity or allergy to sulfobutylether-7-b-cyclodextrin; clopidogrel bisulphate tablet or its excipients; or a positive screen for hepatitis B, hepatitis C, or HIV.
2.4 Study Drugs
MDCO-157, a formulation of clopidogrel complexed with sulphobutylether betacyclodextrin (Captisol®) as an excipient, and clopidogrel (Plavix®) were provided by the study sponsor (The Medicines Company). The MDCO-157 doses chosen for the current clinical study (75, 150, and 300 mg) were based on previous human exposures in Study 104 [19], where MDCO-157 was safe and well tolerated.
2.5 Pharmacodynamics Laboratory Methods
Platelet function assays were performed immediately after venipuncture in a single laboratory by operators blinded to the assigned dosing regimen and to the genetic profile of the study individuals. VASP phosphorylation was measured by flow cytometry (FC-VASP) using a Beckman Coulter FC500 cytometer (Beckman Coulter, Villepinte, France) according to the manufacturer’s instructions and as described in previous studies [17, 18]. For FC-VASP, blood samples were incubated in vitro with ADP and/or prostaglandin E1 (PGE1) before fixation. The VASP platelet reactivity index (PRI) was calculated from the median fluorescence intensity (MFI) of each condition according to the formula: FC-VASP PRI = ((MFI [PGE1]−MFI [PGE1 + ADP])/MFI [PGE1]) × 100. LTA (Model 490-4D, Chrono-Log Corporation, Kordia, the Netherlands), the point-of-care (POC) assay, VerifyNow P2Y12 (VN-P2Y12) assay (Accumetrics, San Diego, CA, USA), and VASP assays were performed simultaneously within 30 min prior to dose administration, 5 (±1), 15 (±2), and 30 (±2) min, and 1 (±5 min), 2 (±5 min), 4 (±5 min), 6 (±5 min), 10 (±10 min), and 24 (±10 min) h after the end of dosing. Residual platelet aggregation (RPA), which corresponds to the level of aggregation curve (%) measured 6 min after 20 µmol/l ADP-induced platelet aggregation, was selected as the platelet aggregation measure because it is thought to better reflect P2Y12 function than other measurements [16]. The relative reduction (RR) in RPA (RR-RPA, %) 6 h post-Loading dose (LD) was defined as (% RPA_6 h)(% RPA_baseline)/(% RPA_baseline) × 100. Pre-specified criteria to define non-evaluable samples were the lack of sufficient signal, hemolysis, platelet-rich plasma (PRP) platelet count <150,000/µl, and unstable baseline. For the VN-P2Y12 assay, samples were run in the same laboratory, according to the device package insert. Results were expressed as platelet reaction units (PRU), which was measured and correlated to RPA at each time point.
2.6 Plasma Concentrations of Clopidogrel, Clopidogrel Carboxylic Acid, and H4-Thiol Active Metabolite
Whole blood was immediately stabilized with 500 mM MPBr (3-methoxyphenacyl bromide) in acetonitrile, and samples were processed to obtain platelet-poor plasma aliquots, and then stored at −80 °C. Clopidogrel, clopidogrel carboxylic acid, and clopidogrel H4-thiol concentrations were determined by a validated liquid chromatography tandem mass spectrometry method. Two bioanalytical assay methods were used, validated and conducted by Advion Bioanalytical Labs/Quintiles Bioservices (Ithaca, NY, USA). The second method was developed and validated for purposes of the present study, with a shift in the clopidogrel concentration range to accommodate higher clopidogrel concentrations in plasma after administration of MDCO-157 infusion doses. Plasma samples were obtained at baseline (within 30 min prior to study drug administration = pre-dose), 5 (±1), 15 (±2), 30 (±2) min, and 1 (±5 min), 2 (±5 min), 4 (±5 min), 6 (±5 min), 10 (±10 min), and 24 (±10 min) h after the end of infusion. An additional PK sample was taken just prior to the end of the MDCO-157 infusion.
2.7 Genetic Analyses
Genomic DNA was extracted from peripheral blood and was genotyped using TaqMan Validated single nucleotide polymorphism (SNP) assays (C_25986767_70) with the 7900HT sequence Detection System (Applied Biosystems, Courtaboeuf, France). All patients were also genotyped for other loss-of-function CYP2C19 variants (*2, *3, *4, *5, *6) using the same method (TaqMan Validated SNP assays: C_27861809_10; C_30634136_10; C_27861810_10; and C_27531918_10, respectively). ‘Ultrarapid’ metabolizers were defined as carriers of the CYP2C19*17 variants, ‘extensive’ as wild-type carriers, ‘intermediate’ as heterozygous carriers of the CP2C19*2 variant, and ‘poor’ as homozygous carriers of the CP2C19*2 or CP2C19*6 variant.
2.8 Study Oversight
The trial was conducted by members of the nonprofit academic research organization ACTION (Allies in Cardiovascular Trials, Initiatives, and Organized Networks; [http://www.action-coeur.org]) and the Department of Pharmacology. Funding was from the Medicines Company (Parsippany, NJ, USA), which was involved in the design and conduct of the study, data collection, analysis of the results, and writing of the manuscript. The trial was designed and the protocol and manuscript were written by the first and last authors, who made the decision to submit the manuscript for publication together with The Medicines Company. The present study was conducted in accordance with the Harmonized Tripartite Guidelines for Good Clinical Practice issued by the International Conference on Harmonization (http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf, 17 October 2011) and with the local laws and regulations for the use of investigational therapeutic agents. The protocol and informed consent forms were reviewed and approved by the ANSM (Agence Nationale de Sécurité du Médicament et des produits de santé) and the Comité de Protection des Personnes prior to screening or enrolling of any study subjects. All subjects provided voluntary informed written consent to participate in the study.
2.9 Statistical and Data Analyses
Enrollment was planned to include approximately 36 subjects, to ensure that approximately 32 subjects would complete all study periods. Subjects who dropped out prior to completing all the study periods were not replaced. Assuming an intra-subject coefficient of variation (% CV) of 40 % for the primary endpoints (E max and AUEC of P2Y12 receptor inhibition), a sample size of 32 subjects was calculated to provide 80 % power to demonstrate the bioequivalence of PD effects between the matched dose level of MDCO-157 and clopidogrel 300 mg using two one-sided tests at a 5 % significance level with the equivalence range of the mean ratio from 75 to 133 %. Descriptive statistics for continuous variables included number of subjects (n), mean, and standard deviation (SD).
The area under the plasma concentration versus time curve (AUC) from the time of administration of the study drugs to 6 h (AUC0–6) of clopidogrel and its metabolites was computed by non-compartmental methods of analysis. PK variables were calculated using standard, non-compartmental methods with the use of the log-linear trapezoidal method (Phoenix WinNonlin, version 6.3, Certara, L.P.). PK parameters assessed were as follows: maximum observed plasma concentration (C max), time to reach C max (t max), area under the curve of the plasma concentration to the last measurable concentration (AUClast), area under the curve of the plasma concentration to infinity (AUCinf), terminal elimination rate constant (λz), half-life (t ½), volume of distribution at steady state (V ss), and clearance (CL).
For PK parameters, geometric mean, standard error of the mean (SEM), and % CV were calculated. Unless otherwise specified, missing data were not imputed and were excluded from the associated analysis. The dose–response relationship between the PD endpoints and treatment groups was graphically depicted as box and whisker plots of E max and AUEC based on all PD data from the three MDCO-157 dose levels. A linear regression analysis was used to summarize and model the dose–response relationship separately for E max and AUEC among all MDCO-157 dose levels, and to estimate a plausible dose level of MDCO-157 matching a clopidogrel dose of 300 mg. Based on PD response, PK data from the active metabolite were used for modeling. The relationship between PD effects (P2Y12 receptor and platelet aggregation inhibition) and plasma concentration of clopidogrel H4-thiol active metabolite at each time point was summarized graphically. The impact of genetic testing results on the PD and PK effects was explored. No statistical adjustment for multiple comparisons was performed.
3 Results
3.1 Subject Disposition
All 37 enrolled subjects were included in the safety population and in the PK population. A summary of the baseline characteristics is shown in Table 1. One subject who received an incomplete dose due to infusion site extravasation was counted twice in the safety and PK MDCO-157 (75 mg) group, according to actual treatment received. The PD population consisted of 36 subjects, with one subject excluded because of sample hemolysis. Subjects without evaluable study drug levels in PK samples or without evaluable PD data were omitted from PK or PD dose groups (Fig. 2).

Disposition of subjects. aSubject did not complete the study after withdrawing consent, with no study drug received from Treatment Period. bSubject did not complete the study due to other reasons than those listed on the e-case report form (eCRF), with no study drug received from Treatment Period 4 onwar. cSubject did not complete the study due to anti-inflammatory treatment taken by the subject, with no study drug received from Treatment Period 2 onward
3.2 Pharmacodynamics
MDCO-157 demonstrated dose-related PD effects across the intravenous 75-, 150-, and 300-mg dose groups that were lower in magnitude than oral clopidogrel 300 mg PD effects as assessed by the VASP assay (primary endpoint) (Table 2). Numerically, mean E max based on VASP decreased with increasing intravenous doses, and was lower for oral clopidogrel 300 mg than for the intravenous 300-mg group (p < 0.001). Mean AUEC for VASP was higher for the intravenous 300 mg dose than for the lower intravenous doses, without increasing between the 75-mg and the 150-mg dose levels. The mean AUEC was approximately twice as high for the clopidogrel 300-mg group as for the intravenous 300-mg group (Fig. 3). Intravenous MDCO-157 doses showed transient and minor PD effects compared with the oral clopidogrel 300-mg dose, as assessed by LTA (data not shown) and the VN-P2Y12® assay. This was in contrast with observations of a persistent platelet inhibition with oral clopidogrel (minimum VN-P2Y12 PRU was 259.1 ± 44.2, 247.8 ± 49.8, 233.3 ± 65.1 for intravenous 75, 150, and 300 mg, respectively) and higher than oral 300-mg administration (168.3 ± 72.0; pairwise p < 0.0001) (Fig. 4). PD obtained with the oral clopidogrel 300-mg dose were as expected based on known product characteristics and consistent with the scientific literature [3].

Box and whisker plot of VASP AUEC versus dose. AUEC area under the effect of P2Y12 receptor inhibition time curve, IV intravenous, VASP vasodilator-stimulated phosphoprotein phosphorylation

Pharmacodynamic response at each time point by treatment: VerifyNow® PRU results and PRU % inhibition (upper PRU results) and (lower PRU % inhibition). hr hours, IV intravenous, PRU P2Y12 reaction units, SE standard error
3.3 Pharmacodynamic Response versus CYP2C19 Polymorphism
Of 37 subjects, nine were predicted to be poor responders based on CYP2C19 metabolizer genotypes. Based on PK analysis, median AUC for the active H4-thiol metabolite appeared to be lowest in subjects who were poor metabolizers and highest in ultra-metabolizers (data not shown). PD analysis of VASP AUEC versus CYP2C19 polymorphism was performed; median AUEC appeared to be lower in the poor and intermediate metabolizers than in the extensive and ultra-metabolizers (Fig. 5).

Box and whisker plot of VASP AUEC by CYP2C19 polymorphism, oral clopidogrel 300 mg. AUEC area under the effect of P2Y12 receptor inhibition time curve, EM extensive metabolizer, IM intermediate metabolizer, PM poor metabolizer, RM ultrarapid metabolizer, VASP vasodilator-stimulated phosphoprotein phosphorylation
3.4 Pharmacokinetics
PK variables for clopidogrel, the inactive clopidogrel carboxylic acid metabolite, and the active clopidogrel H4-thiol metabolite were compared for intravenous MDCO-157 doses of 75, 150, and 300 mg, and oral clopidogrel 300 mg. The mean (±SD) C max for clopidogrel increased with intravenous MDCO-157 dose. The increase in C max was dose proportional, with a slope in log scale of 0.998 (90 % confidence interval [CI] 0.817–1.18). Similarly, mean AUC increased with dose but was slightly less than dose proportional, with a slope in log scale of 0.863 (90 % CI 0.768–0.958). After intravenous MDCO-157 dosing, clopidogrel showed a high clearance, short half-life, and higher C max and AUC than that of the oral clopidogrel 300-mg dose (Table 3).
The mean (±SD) C max for the H4-thiol active metabolite increased with intravenous MDCO-157 dose. The increase in C max was slightly less than dose proportional, with a slope in log scale of 0.799 (90 % CI 0.691–0.907). AUC increased with IV dose, with the increase in AUCinf slightly less than dose proportional. After intravenous MDCO-157 300 mg, the H4-thiol active metabolite had lower C max and AUC than that of the oral clopidogrel 300-mg dose (Table 3).
3.5 Pharmacodynamics and Pharmacokinetics
PD responses across intravenous MDCO-157 dose levels were determined to be insufficient for modeling of dose response to estimate the intravenous MDCO-157 dose matching oral clopidogrel 300 mg. As a result, PK data were used for modeling and prediction of the MDCO-157 matching dose.
The plasma exposure for the active H4-thiol metabolite was lower than that for the inactive carboxylic acid. The mean AUCinf ratio of the active metabolite to the inactive metabolite was approximately 0.00031, 0.00028, and 0.00023 for the intravenous 75, 150, and 300-mg groups, and was 0.00084 for the oral 300-mg group. The ratio for the oral 300-mg group was approximately 3.65 times higher than that of the intravenous 300-mg group. Based on a linear model of dose proportionality of C max and AUC, the intravenous MDCO-157 dose that would be expected to produce similar plasma exposure of the H4-thiol metabolite compared with clopidogrel 300 mg was estimated to be 500–900 mg.
The hysteresis plots of mean VASP-PRI results versus mean H4-thiol active metabolite concentrations are presented in Fig. 6. For the oral clopidogrel 300-mg dose, there was a time delay between the measurement of the active metabolite and the VASP-PRI response. For the intravenous MDCO-157 dose levels, the time delay and the inhibition of the VASP-PRI was not as notable as it was for the oral clopidogrel group.

Hysteresis plot of mean VASP versus mean concentration of H4-thiol active metabolite. IV intravenous, PRI platelet reactivity index, PRU P2Y12 reaction units, VASP vasodilator-stimulated phosphoprotein phosphorylation, arrows indicate the direction of the hysteresis
For the oral clopidogrel 300-mg dose, VASP AUEC was well correlated with C max for the active H4-thiol metabolite. For the intravenous MDCO-157 dose levels, C max values for the active metabolite did not appear to reach a level that could cause marked changes in VASP AUEC, except for one subject (Fig. 7). LTA results (RPA and Maximum Platelet Aggregation (MPA) with 5 and 20 µM ADP) showed poor correlation between AUEC and C max or AUEC and AUC for the active H4-thiol metabolite. VN-P2Y12-PRU results were more consistent with those based on VASP assessments (data not shown).

Correlation between H4-thiol maximum observed plasma concentration (C max) and vasodilator-stimulated phosphoprotein (VASP) area under the effect of P2Y12 receptor inhibition time curve (AUEC). IV intravenous
3.6 Safety Results
Safety findings after the administration of intravenous MDCO-157 doses at 75, 150, and 300 mg were consistent with the safety and tolerability profile of MDCO-157, as previously reported. No deaths, serious adverse events (SAEs), or adverse events (AEs) leading to discontinuation of study drug were reported among study subjects. After the administration of intravenous MDCO-157 at 300 mg (the highest MDCO-157 dose), the most commonly reported treatment-emergent AEs (TEAEs) were infusion-site pain (28 of 35 subjects; 80.0 %), headache (6/35; 17.1 %), dizziness (5/35; 14.3 %), and vessel puncture site hematoma (4/35; 11.4 %). After the administration of oral clopidogrel 300 mg, the most commonly reported TEAE was headache (4/36; 11.1 %). The majority of study subjects had no notable findings with respect to laboratory results, vital signs, or ECGs (see Table 4).
4 Discussion
The AMPHORE study first demonstrates that when administered in intravenous doses from 75 up to 300 mg, MDCO-157, an intravenous formulation of clopidogrel bisulfate, produced PD effects transient and minor for the majority of study subjects, which were lower in magnitude than the PD effects observed with the administration of oral clopidogrel 300 mg. The PK and PD analyses of clopidogrel 300 mg were consistent with its known PK and PD profiles [3, 21, 22]. The second important finding of AMPHORE is the dose-proportional kinetics for clopidogrel, the carboxylic acid and the H4-thiol active metabolites after intravenous administration of MDCO-157. Third, the AUC of clopidogrel was very high with MDCO-157 compared with oral administration, with a total body clearance higher than hepatic blood flow (~90 l/h/70 kg), reflecting extrahepatic clearance, which may result in decreased hepatic metabolism via this route of administration. Finally, the inactive carboxylic acid metabolite showed higher plasma exposure than the parent clopidogrel.
The findings of the AMPHORE study differ substantially from the PRISM-104 study, where the administration of PM-103 produced a rapid, persistent, and dose-related inhibition of platelet aggregation [19]. The onset of the antiplatelet effect paralleled the appearance in plasma of the clopidogrel thiol active metabolite. But the lack of PK/PD dose-dependent relationship was an intriguing finding attributed to the lability of the H4-thiol metabolite, further questioning the accuracy of the bioanalytical assay used in the PRISM-104 study. Differences in the sulphobutylether betacyclodextrin formulation and longer time delay from blood sampling to biological assay may also have played a role. Sulphobutylether betacyclodextrin was used in 100 mM phosphate buffer, pH 6.5 in PRISM-104 and in 10 mM phosphate buffer, pH 6.0 in AMPHORE. Finally, AMPHORE was designed and executed according to the FDA requirements for a phase I study in normal healthy volunteers, a guarantee of reliability and accuracy. In particular, multiple PD assays were run at each time point and duplicated to ensure that the correlation between PK and PD measures were reliable.
In AMPHORE, the P2Y12 inhibition effects of MDCO-157 were assessed using VASP, VerifyNow®, or LTA. VASP and VerifyNow® were found to correlate with the plasma exposure of the H4-thiol active metabolite (C max and AUC) as opposed to RPA and MPA using LTA. VASP and VerifyNow® results are known to be more specific assessments of P2Y12 inhibition than LTA, a finding that is consistent with AMPHORE. The PK/PD analysis of MDCO-157 in AMPHORE also indicates that there might be a threshold for the active metabolite to cause stronger PD responses than previously observed. The plasma levels of the active H4-thiol metabolite after intravenous administration of MDCO-157 appear to be lower than the threshold suggested by the results of PK/PD analysis [3, 23]. This threshold was determined to be 24 ng/ml when PK and PD responses to high or standard clopidogrel loading doses were modeled according to CYP2C19*2 allele carriage [3], a threshold that fits with our data. Indeed, the hysteresis curve in Fig. 6 suggests a threshold effect of H4-thiol between concentration 15 and 23 ng/ml to obtain an irreversible inhibition of the P2Y12.
The MDCO-157 intravenous dose expected to produce similar plasma exposure of the H4-thiol metabolite compared with clopidogrel 300 mg was estimated to be 500–900 mg. This suggests that higher doses and/or doses with longer infusion may have been needed to reach a sufficient threshold of active metabolite generation. However, the partition ratio between the H4-thiol metabolite and the carboxylic acid metabolite after intravenous MDCO-157 at 300 mg, which was calculated using the corresponding AUC values, was found to be only 28 % of that calculated for oral clopidogrel 300 mg. This further indicates that the 300-mg dose of MDCO-157 may not have been sufficient to overcome the poor PD response. Based on these assumptions, an oral dose of 300 mg would match an intravenous dose of 600 mg, a value consistent with our calculations.
Three possible mechanisms may be proposed to explain this finding, which reflects the lower plasma exposure for the H4-thiol active metabolite after MDCO-157 dosing compared with oral clopidogrel. One could be the oxidation of clopidogrel by intestinal CYP450 enzymes to form the thiolactone metabolite, which would then be delivered through the portal vein to the liver for further oxidation to the active metabolite. Another possible mechanism could be complex formation between sulphobutylether betacyclodextrin (in the MDCO-157 formulation) and clopidogrel, which might reduce the diffusion rate of clopidogrel to hepatocytes for metabolism and/or might increase the renal clearance of clopidogrel. In vitro data support this hypothesis (not shown). Finally, arterial hepatic blood flow represents 20–25 % of the total liver blood supply, 75–80 % being supplied by the portal vein. The amount of clopidogrel and its esterase-dependent carboxylic acid derivative delivered to the liver would therefore represent about a quarter of the amount delivered to the liver after oral administration. Absorption of clopidogrel after oral administration is at least 50 %. Based on these assumptions, an oral dose of 300 mg would match an intravenous dose of 600 mg, a value consistent with our calculations.
A possible effect of the CYP2C19 polymorphism can be excluded in the AMPHORE study. The PD effects and the plasma exposure of the active metabolite after clopidogrel were highly variable among subjects. Genotyping of study subjects and analysis of the CYP2C19 genotype categories (poor, intermediate, extensive, and ultrarapid) showed an apparent correlation between metabolizer phenotype and the plasma exposure of the H4-thiol active metabolite, as well as VASP and PRU PD results. These findings were expected, and the magnitude of effect of the CYP2C19 polymorphism was similar to that observed in the previous study, and considered only as one possible factor contributing to the significant inter-individual variability in clopidogrel PK and PD [3, 24–26].
In conclusion, early and profound platelet P2Y12 receptor inhibition is an attractive approach that is recommended to improve clinical outcome of high-risk PCI patients [27]. The MDCO-157 compound was developed to provide such a strategy and to allow an easy transition from intravenous to oral administration using clopidogrel, the most widely used P2Y12 inhibitor in ACS patients worldwide. Although safety and tolerance were good in normal healthy volunteers, there was a lack of sufficient platelet inhibition when given at doses up to 300 mg due to insufficient threshold of active metabolites generation. The ratio of hepatic arterial-to-portal venous blood flow was the most plausible explanation for our results and whether a higher dose would overcome this limitation is unknown. Finally, MDCO-157 at these doses cannot provide an alternative to oral clopidogrel for clinical use. The use of higher doses of MDCO-157 may face volume issues for intravenous use.
References
Savi P, Labouret C, Delesque N, Guette F, Lupker J, Herbert JM. P2Y(12), a new platelet ADP receptor, target of clopidogrel. Biochem Biophys Res Commun. 2001;283:379–83.
Savi P, Pereillo JM, Uzabiaga MF, et al. Identification and biological activity of the active metabolite of clopidogrel. Thromb Haemost. 2000;84:891–6.
Collet J-P, Hulot J-S, Anzaha G, et al. High doses of clopidogrel to overcome genetic resistance: the randomized crossover CLOVIS-2 (Clopidogrel and Response Variability Investigation Study 2). JACC Cardiovasc Interv. 2011;4:392–402.
Bellemain-Appaix A, O’Connor SA, Silvain J, et al. Association of clopidogrel pretreatment with mortality, cardiovascular events, and major bleeding among patients undergoing percutaneous coronary intervention: a systematic review and meta-analysis. JAMA. 2012;308:2507–16.
Bellemain-Appaix A, Brieger D, Beygui F, et al. New P2Y12 inhibitors versus clopidogrel in percutaneous coronary intervention: a meta-analysis. J Am Coll Cardiol. 2010;56(19):1542–51.
Montalescot G, Wiviott SD, Braunwald E, et al. Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST-elevation myocardial infarction (TRITON-TIMI 38): double-blind, randomised controlled trial. Lancet. 2009;373(9665):723–31.
Steg PG, James S, Harrington RA, et al. Ticagrelor versus clopidogrel in patients with st-elevation acute coronary syndromes intended for reperfusion with primary percutaneous coronary intervention: a platelet inhibition and patient outcomes (PLATO) trial subgroup analysis. Circulation. 2010;122(21):2131–41.
Montalescot G, van’t Hof AW, Lapostolle F, et al. Prehospital ticagrelor in ST-segment elevation myocardial infarction. N Engl J Med. 2014;371:1016–27.
Alexopoulos D, Xanthopoulou I, Gkizas V, et al. Randomized assessment of ticagrelor versus prasugrel antiplatelet effects in patients with ST-segment-elevation myocardial infarction. Circ Cardiovasc Interv. 2012;5:797–804.
Franchi F, Angiolillo DJ. Novel antiplatelet agents in acute coronary syndrome. Nat Rev Cardiol. 2015;12:30–47.
Steg PG, Bhatt DL, Hamm CW, et al. Effect of cangrelor on periprocedural outcomes in percutaneous coronary interventions: a pooled analysis of patient-level data. Lancet. 2013;382:1981–92.
European Medicines Agency. http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/003773/WC500180855.pdf. 2015.
Anon. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm452172.htm.
Bhatt DL, Stone GW, Mahaffey KW, et al. Effect of platelet inhibition with cangrelor during PCI on ischemic events. N Engl J Med. 2013;368:1303–13.
Rollini F, Franchi F, Tello-Montoliu A, et al. Pharmacodynamic effects of cangrelor on platelet P2Y12 receptor-mediated signaling in prasugrel-treated patients. JACC Cardiovasc Interv. 2014;7:426–34.
Angiolillo DJ, Schneider DJ, Bhatt DL, et al. Pharmacodynamic effects of cangrelor and clopidogrel: the platelet function substudy from the cangrelor versus standard therapy to achieve optimal management of platelet inhibition (CHAMPION) trials. J Thromb Thrombolysis. 2012;34:44–55.
Schneider DJ, Agarwal Z, Seecheran N, Keating FK, Gogo P. Pharmacodynamic effects during the transition between cangrelor and ticagrelor. JACC Cardiovasc Interv. 2014;7:435–42.
Schneider DJ, Seecheran N, Raza SS, Keating FK, Gogo P. Pharmacodynamic effects during the transition between cangrelor and prasugrel. Coron Artery Dis. 2015;26:42–8.
Cushing DJ, Souney PF, Cooper WD, et al. Pharmacokinetics and platelet aggregation inhibitory effects of a novel intravenous formulation of clopidogrel in humans: effects of intravenous clopidogrel. Clin Exp Pharmacol Physiol. 2012;39:3–8.
Williams EJ. Experimental designs balanced for the estimation of residual effects of treatments. Aust J Sci Res 1949;2(3):149–168
Montalescot G, Sideris G, Meuleman C, et al. A randomized comparison of high clopidogrel loading doses in patients with non-ST-segment elevation acute coronary syndromes: the ALBION (Assessment of the Best Loading Dose of Clopidogrel to Blunt Platelet Activation, Inflammation and Ongoing Necrosis) trial. J Am Coll Cardiol. 2006;48:931–8.
Collet JP, Silvain J, Landivier A, et al. Dose-effect of clopidogrel re-loading in patients already on 75 mg maintenance dose: the RELOAD study. Circulation. 2008;118:1225–33.
Simon N, Finzi J, Cayla G, Montalescot G, Collet JP, Hulot JS. Omeprazole, pantoprazole, and CYP2C19 effects on clopidogrel pharmacokinetic-pharmacodynamic relationships in stable coronary artery disease patients. Eur J Clin Pharmacol 2015;71(9):1059–66
Pena A, Collet JP, Hulot JS, et al. Can we override clopidogrel resistance? Circulation. 2009;119(21):2854–8.
Hulot JS, Collet JP, Cayla G, et al. CYP2C19 but not PON1 genetic variants influence clopidogrel pharmacokinetics, pharmacodynamics and clinical efficacy in post-myocardial infarction patients. Circ Cardiovasc Interv. 2011;4:422–8.
Frelinger AL 3rd, Bhatt DL, Lee RD, et al. Clopidogrel pharmacokinetics and pharmacodynamics vary widely despite exclusion or control of polymorphisms (CYP2C19, ABCB1, PON1), noncompliance, diet, smoking, co-medications (including proton pump inhibitors), and pre-existent variability in platelet function. J Am Coll Cardiol. 2013;61:872–9.
Windecker S, Alfonso F, Collet J-P, et al. 2014 ESC/EACTS guidelines on myocardial revascularization: the task force on myocardial revascularization of the ESC/EACTS Developed with the special contribution of the EAPCI. Eur Heart J. 2014;35:2541–619.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Montalescot reports receiving consulting fees from Acuitude, Amgen, AstraZeneca, Bayer, Berlin Chimie AG, Boehringer Ingelheim, Bristol-Myers-Squibb, Brigham Women’s Hospital, Cardiovascular Research Foundation, CME resources, Conway, Daiichi-Sankyo, Eli-Lilly, Europa, Evidera, GLG, Hopitaux Universitaires Genève, Lead-Up, McKinsey & Company, Medcon International, Menarini, Medtronic, MSD, Pfizer, Sanofi-Aventis, Stentys, The Medicines Company, TIMI Study Group, Universität Basel, WebMD, Williams & Connolly, and Zoll Medical; and grant support from ADIR, Amgen, AstraZeneca, Bristol-Myers-Squibb, Celladon, Daiichi-Sankyo, Eli-Lilly, Fédération Française de Cardiologie, Gilead, ICAN, Janssen-Cilag, Pfizer, Recor, Sanofi-Aventis, Stentys. Dr. Silvain reports receiving research grants to the institution from Boehringer-Ingelheim, Daiichi-Sankyo, Eli Lilly, BRAHMS, and Sanofi-Aventis, Fédération Française de Cardiologie and Société Française de Cardiologie, INSERM; consultant fees from Daiichi-Sankyo, Eli Lilly, AstraZeneca, and The Medicines Company; and lecture fees from AstraZeneca, Cordis, Daiichi-Sankyo, Eli Lilly, Iroko Cardio, and STENTYS. Jayne Prats is an employee of The Medicines Company; Ming-yi Hu and Kan He were employees of The Medicines Company. Dr. Collet has received research grants from Bristol-Myers Squibb, Sanofi-Aventis, Eli Lilly, Guerbet Medical, Medtronic, Boston Scientific, Cordis, Stago, Centocor, Fondation de France, INSERM, Fédération Française de Cardiologie, and Société Française de Cardiologie; consulting fees from Sanofi-Aventis, Eli Lilly, and Bristol-Myers Squibb; and lecture fees from Bristol-Myers Squibb, Sanofi-Aventis, and Eli Lilly. Dr. Hulot discloses the following relationships: Data Monitoring Committees: Assistance Publique Hôpitaux de Paris; Honoraria: American College of Cardiology Foundation (Lecturer), Eli Lilly (Steering Committee), Servier (Lecturer); Consulting fees: Daiichi Sankyo (consultant), Imaxio (consultant); Research Grants: Bayer Pharma, Celladon, Cellectis Stem Cells, French Ministry of Health (AOM09191), Institute for Cardiometabolism and Nutrition Foundation, NIH/NHLBI (R01 HL113497), NIH/NGRI (1U01HG006380). The other authors declare no conflicts of interest.
Additional information
N. Nicolas: Deceased.
Rights and permissions
About this article

Cite this article
Collet, JP., Funck-Brentano, C., Prats, J. et al. Intravenous Clopidogrel (MDCO-157) Compared with Oral Clopidogrel: The Randomized Cross-Over AMPHORE Study. Am J Cardiovasc Drugs 16, 43–53 (2016). https://doi.org/10.1007/s40256-015-0145-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-015-0145-0