
Abstract
Background
The management of schizophrenia has seen significant strides over the last few decades, due to the increasing availability of a number of antipsychotics. Yet, the diminished efficacy in relation to the negative and cognitive symptoms of schizophrenia, and the disturbing adverse reactions associated with the current antipsychotics, reflect the need for better molecules targeting unexplored pathways.
Purpose
To review the salient features of the recently approved antipsychotics; namely, iloperidone, asenapine, lurasidone and blonanserin.
Methods
We discuss the advantages, limitations and place in modern pharmacotherapy of each of these drugs. In addition, we briefly highlight the new targets that are being explored.
Results
Promising strategies include modulation of the glutamatergic and GABAergic pathways, as well as cholinergic systems.
Conclusions
Although regulatory bodies have approved only a handful of antipsychotics in recent years, the wide spectrum of targets that are being explored could eventually bring out antipsychotics with improved efficacy and acceptability, as well as the potential to revolutionize psychiatric practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Schizophrenia is a severe form of mental illness which affects millions of people worldwide, affecting mostly the productive age group. The disease causes considerable morbidity and places a major financial burden on the society. Although there are various forms of the illness, the symptoms of the disease can be broadly classified into positive symptoms (delusions, hallucinations), negative symptoms (avolition, affective flattening, impoverishment of speech) and cognitive symptoms(attention deficits, impaired memory). Patients with schizophrenia are at almost ten times greater risk of suicidal behaviour compared to the general population [1]. Since the introduction of chlorpromazine, the first antipsychotic in 1952, more than 50 kinds of antipsychotics have been marketed for schizophrenia therapy worldwide. Antipsychotics have revolutionized psychiatric practice by offering patients the opportunity to receive domiciliary treatment, thereby removing the stigma of being institutionalized in mental asylums. These drugs have remarkably changed the morbidity associated with the illness. Yet, more than 30 % of patients relapse after 1 year of treatment with antipsychotics [2]. Most patients on antipsychotics also suffer from several adverse drug reactions which takes a heavy toll on the quality of their life. In addition, most antipsychotics have few or no effects on the cognitive and negative symptoms of schizophrenia as compared to their control of positive symptoms. This has led to an active search for better drug molecules with favorable safety profiles and improved efficacy.
The pathophysiology of this disease remains an enigma, though it has been recognised more than a hundred and fifty years ago. Traditionally overactivity of the dopaminergic neurons in the basal forebrain has been described as the major pathogenic event, while subsequent research recognized the importance of targeting the serotonergic pathway. Recent genome wide association studies have confirmed the fact that schizophrenia is a polygenic disease [3]. This review focuses on antipsychotics that have been approved by the regulatory agencies in the last 4 years and upcoming drug molecules that are being explored to target this devastating mental disorder in the light of recent advances in the field .
Iloperidone
Iloperidone is a piperidinyl benzisoxazole derivative that is structurally akin to ziprasidone and paliperidone [4]. The drug was approved by the US FDA (Food and Drug Administration) in May 2009 for the treatment of acute schizophrenia. Its pharmacological actions are brought about by its high affinity for D2 and 5HT2A receptors. Iloperidone also has high affinity for dopamine D3 receptors, serotonin 5-HT2A receptors and α1 adrenergic receptors. Molecular imaging techniques such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT) have enabled us to understand the chemical changes in the brain of schizophrenia patients. The synthesis and availability of neurotransmitters and the neurotransmitter receptors occupancy by the antipsychotics can be accurately gauged by these studies. Using radioactive tracers tagged to the antipsychotic molecule, the binding affinity of iloperidone to the different types of receptors can be determined [5]. A PET study done in schizophrenic patients showed that iloperidone at a dose of 8–12 mg showed maximal D2 receptor occupancy in the caudate nucleus and putamen [6]. It has the highest affinity for α adrenergic receptors next only to aripiprazole, leading to an increased incidence of orthostatic hypotension and syncope, but a potential advantage of improved cognition. Iloperidone is one of the antipsychotics for which genetic polymorphisms have been shown to predict the response and the development of side effects like QT prolongation [7].
Pharmacokinetics
Iloperidone is well absorbed orally with a bioavailability of 96 %. Being metabolised by the CYP2D6 enzyme system, the elimination half-life (t1/2) of iloperidone ranges from 18 h for extensive CYP2D6 metabolizers to 33 h for poor metabolizers [8]. Hepatic metabolism involves O-dealkylation, hydroxylation and decarboxylation/reduction mediated by the cytochrome enzyme system. It is recommended that the dose of iloperidone be reduced by 50 % when strong inhibitors of CYP2D6 (fluoxetine) or CYP3A4 (Ketoconazole) are co-administered. Since SSRIs are frequently prescribed along with antipsychotics, this interaction becomes particularly important in clinical settings. The two major metabolites of iloperidone are P95 and P88 [9]. The therapeutic concentration range for iloperidone is 5–10 ng/ml [10].
Efficacy
Four Phase III randomised double-blind, placebo and active controlled trials led to the approval of iloperidone for the acute treatment of schizophrenia. The initial three studies [11], were conducted for 6 weeks and used either risperidone or haloperidol as the active comparators. Patients diagnosed with schizophrenia or schizoaffective disorder with a PANSS-T [positive and negative syndrome scale, total score] score of ≥60 were included and started on iloperidone in doses of 4, 8 and 12 mg per day. The dose of haloperidol was 15 mg/day and that of risperidone was 4–8 mg/day. The dose of iloperidone was gradually titrated over 1 week to achieve therapeutic concentrations whereas the dose of haloperidol or risperidone was fixed. In all the three studies iloperidone showed significant improvements in the PANSS scores from the baseline value, when compared to placebo.
To overcome the problem of dosage titration schedules, the fourth study administered iloperidone, ziprasidone and placebo in a similar dosage schedule [12]. The dose of all three were escalated over 1 week to reach a fixed dose of iloperidone 24 mg daily, ziprasidone 160 mg daily and placebo twice daily. After 3 weeks of fixed dose schedules, patients were assessed by PANSS and BPRS [Brief Psychiatric Rating Scale] scores, in which the iloperidone group was found to be superior when compared to placebo.
Pooled analysis of these four phase III trials showed that treatment with iloperidone either 10–16 mg or 20–24 mg resulted in significant improvements BPRS and PANSS scores when compared with the placebo. It also showed significant improvements across all dimensions of psychopathology commonly associated with schizophrenia: positive, negative, cognitive, hostility/excitement and mood symptoms. The 10–16 mg group showed significant improvements in all the five aspects of PANSS scores whereas the 20–24 mg group failed to show improvements in depression and negative symptoms. This difference may be due to the small sample size in the higher dose group [13].
A trial which has investigated the long term efficacy of iloperidone in schizophrenia for 52 weeks in comparison to haloperidol did not reveal any significant difference between the two drugs in the primary outcome measure, which was the time to relapse while on treatment. The subjects received either iloperidone 4–16 mg daily or haloperidol 5–20 mg daily. Results showed that patients in both the groups showed similar discontinuation and relapse rates [14]. The drug has not been studied in pregnant women, children and elderly.
Safety & tolerability
In general, iloperidone was well tolerated with a lesser incidence of treatment-emergent adverse events. Post hoc analysis of all the phase III trials showed that the incidence of adverse events were >5 % in any treatment group. Most common adverse event associated with the use of iloperidone was dizziness seen at a dosage of 20–24 mg. Somnolence, dry mouth, dyspepsia and orthostatic hypotension were the other adverse events. Clinically significant weight gain comparable to that of risperidone has been reported with the use of iloperidone 20–24 mg (13 %). [15]. Mild increases in prolactin levels have been reported with the use of iloperidone.
The incidence of extrapyramidal symptoms is particularly less with the use of iloperidone, which has been attributed to its higher affinity for D2 and 5HT-2A receptors in the limbic tract than nigrostriatal tract leading to improved antipsychotic efficacy and reduced risk of extrapyramidal symptoms. Studies have shown overall lower rates of extrapyramidal symptoms and worsening akathisia when compared to risperidone. Paradoxically, the incidence of orthostatic hypotension is higher in the dose range of 10–16 mg daily than with 20–24 mg daily. The dose of iloperidone is titrated over one week to reduce the complications of orthostatic hypotension and syncope. The drug should be avoided in elderly patients with dementia related psychoses.
Dose related QT prolongation is extensively reported with iloperidone. The drug prolongs cardiac repolarization by acting as a potent HERG blocker [16]. A greater risk of developing QT prolongation occurs in those patients with hypokalemia, hypomagnesemia, bradycardia and those who already are on QT prolonging drugs. This adverse effect of iloperidone has been positively linked with certain genetic polymorphisms which are discussed below.
Pharmacogenetics of Iloperidone
Genome-wide association studies done till now have shown significant association of genetic polymorphisms with the therapeutic response as well as the development of side effects (Table 1). A study has identified six SNPs which are associated with iloperidone response. SNPs in NPAS3 (Neuronal PAS domain protein 3 gene), XKR4 (Kell blood group complex subunit-related family, member 4 gene), TNR (tenascin-R gene), GRIA 4 (Glutamate receptor, ionotropic, AMPA 4 gene), GFRA2 (Glial cell line–derived neurotropic factor receptor-α2) and HTR7 (between NUDT9P1 pseudogene and serotonin receptor gene). The presence of a specific polymorphic genotype resulted in a greater improvement in the BPRS scores between 25 % and 68 % more than the mean response of the iloperidone group [17]. When analysed as a group, these six genotypes influenced the clinical response to iloperidone. Out of the possible 64 combination groups, 46 combinations were observed in the iloperidone treated group. Patients were divided into 4 groups based on the number of genotypes associated with enhanced response. Group 1 contained patients with 0 to 2 genotypes associated with enhanced response. Groups 2, 3 and 4 had 3, 4 and >5 genotypes respectively. The probability of >20 % improvement for each of the groups was 10.2 %, 31.8 %, 57.3 % and 81.5 % respectively. The group which carried all six genotypes had the highest likelihood of response(92 %) to iloperidone [18]
.
Genome wide association studies have identified six genes implicated in the development of QT prolongation. Genetic polymorphisms have been identified in the CERKL (Ceramide kinase like), SLCO3A1 (solute carrier organic anion transporter family, member 3A1), BRUNOL4 (Bruno like 4), NRG3(Neuroregulin 3), NUBPL(nucleotide binding protein-like) and PALLD(palladin, cytoskeletal associated protein) genes [19]. Although various polymorphisms have been linked with the efficacy of iloperidone, there is still a definite need for more evidence from prospective studies before these tests are recommended in routine clinical care.
Role of iloperidone in modern practice
Iloperidone has been approved for the treatment of schizophrenia in adults. Its lesser predilection to develop EPS may make it a better alternative in those patients who develop risperidone induced EPS. Among the new atypical antipsychotics, iloperidone is the only drug that has been associated with genetic polymorphisms. Potential disadvantages include the need to titrate the dose over a week to reduce the incidence of orthostatic hypotension. This process of dosage administration may be cumbersome for certain patients and reduce their adherence .QT prolongation is a significant problem that should be kept in mind before prescribing the drug. The efficacy of iloperidone appears to be similar to the currently available antipsychotics lacking a clear benefit over them.
Asenapine
Asenapine is a new atypical antipsychotic that has been developed as a structural modification of the atypical antidepressant mianserin [20]. The drug has an antagonistic action at serotonin receptors (5HT2A, 5HT2B, 5HT2C, 5HT6 and 5 HT7), adrenergic receptors (α1A, α2A, α2B, α2C) dopaminergic receptors (D3 and D4) and histamine receptors, but no action on the muscarinic receptors [21]. Its antipsychotic action is believed to be brought out largely by its high affinity for the 5HT2A receptor. The lack of affinity for muscarinic receptors and the binding to 5HT2A receptors which leads to dopamine activity in the prefrontal cortex is claimed to translate into improved response, especially in those with cognitive and negative symptoms of schizophrenia [22]. A PET study demonstrated that asenapine had significant occupancy of the dopamine receptors with a good correlation obtained between plasma levels of asenapine and D2 receptor occupancy. Asenapine at a dose of 5 mg BD showed 75 % D2 receptor occupancy and at a dose of 10 mg BD showed 85 % D2 receptor occupancy [23]. The drug is currently approved by the US FDA and European Medical Agency (EMA) for the acute and maintenance treatment of schizophrenia as well as for the acute treatment of manic or mixed episodes associated with bipolar I disorder with or without psychotic features as monotherapy or adjunctive medication along with lithium or valproate [24].
Pharmacokinetics
Asenapine is metabolized by CYP1A2. So one should exercise caution when using CYP1A2 inducers (carbamazepine or rifampin) or CYP1A2 inhibitors (fluvoxamine, ciprofloxacin, ketoconazole). Being a weak inhibitor of CYP2D6, concomitant use of CYP2D6 substrates may cause toxicity. The poor bioavailability of asenapine has led to the development of a sublingual formulation that has a reasonable bioavailability [24]. The therapeutic concentration range of asenapine is 3–5 ng/ml [25] (Table 2)
.
Efficacy
Four pivotal 6 week randomized double blind placebo controlled studies which included a positive comparator were done to evaluate the efficacy of asenapine in schizophrenia. In a phase III randomized controlled trial which included 448 patients from 43 sites, asenapine was compared with haloperidol and placebo [26]. Patients were randomized to receive asenapine 5 mg twice daily, 10 mg twice daily, haloperidol 4 mg twice daily or placebo. The PANSS positive subscale score did show that asenapine 5 mg was superior to placebo while the same beneficial effect could not be observed in the PANSS negative subscale score with any of the medication. In a meta-analysis of the trials that evaluated asenapine for schizophrenia, asenapine was found to have a significantly greater change in the PANSS score compared to placebo [27].
Safety and tolerability
The most common adverse effects that one would encounter when prescribing asenapine are somnolence, dizziness, weight gain and extrapyramidal symptoms. The lesser affinity for D2 receptors offers a benefit by decreasing the risk of EPS and hyperprolactinemia as seen with potent drugs such as haloperidol. Although there is a risk of weight gain with asenapine, it is lesser than that associated with olanzapine. This has been attributed to the lower binding affinity of asenapine for the histamine receptor (H1) as compared to olanzapine and quetiapine which have strong H1 binding affinity and thereby carry a greater risk of weight gain. Elevated fasting glucose and cholesterol elevation may occur in some patients as with other atypical antipsychotics [24].
Role of asenapine in psychiatric practice
Asenapine has been approved for the treatment of acute schizophrenia as well as manic episodes in bipolar disorder. Although asenapine may not occupy a unique role in the treatment of these conditions, its sublingual formulation may offer a specific advantage in a subset of patients who cannot swallow pills due to an esophageal stricture, gastric bypass surgery or a general unwillingness to swallow pills. The rapid onset of action with the sublingual route may also offer a potential advantage in those patients requiring rapid relief of symptoms. Asenapine may be a poor choice for those patients with coexisting depressive symptoms since the drug does not have a significant effect in relieving depressive symptoms.
Lurasidone
Lurasidone, a benzothiazol derivative, is the latest atypical antipsychotic that was approved by the US FDA in 2010. It has a strong antagonistic action at D2 and 5HT2A receptors. It also has a high affinity for 5HT7 and 5 HT1A receptors and a moderate affinity for adrenergic α2C receptors. The drug has no action on histaminergic and muscarinic receptors [28–30]. A PET study done in conscious marmosets showed that lurasidone preferentially binds to D2/D3 receptors rather than 5-HT2A receptors. D2/D3 receptor occupancy was found to be greater than 80 % and correlated with the plasma lurasidone levels [31].
Pharmacokinetics
Lurasidone is well absorbed orally to reach a steady state plasma concentration within 7 days. Since the AUC and Cmax are increased by almost three fold when consumed with food, the current recommendation is to take lurasidone concomitantly with atleast 350 calories of food. The drug should not be co administered with CYP3A4 inducers or inhibitors as it is metabolized by the enzyme CYP3A4. The drug is not a substrate of P-gp [P –glycoprotein]. Drug modifications are necessary in those individuals with impaired hepatic and renal function. There does not appear to be any difference in metabolism with respect to age with the elderly requiring the same dose as normal adults [32].
Efficacy
Prior to approval, as many as eight controlled trials were done to show its efficacy using end points such as the PANSS score, BPRS(Brief Psychiatric Rating Scale) score, CGI –S(Clinical Global Impression-Severity scale) and PANSS subscale scores. Lurasidone at a dose of 80 mg/d showed efficacy on all measures. A pooled analysis which was done from four short term double blind, placebo controlled trials revealed that lurasidone at a dose of 40–120 mg/d was better than placebo in improving all PANSS factor scores such as positive symptoms, negative symptoms, disorganized thought, hostility, and depression/anxiety. Clinical trials have failed to prove any dose response relationship with lurasidone and the 120 mg dose does not seem to have any superiority over the 40 or 80 mg/d doses. Based on these trial results, the drug has been approved by the USFDA with a starting dose of 40 mg/d and a maximum dose of 80 mg/d. The improvements in the different scores were observed within 3 to 7 days of starting lurasidone [33, 34].
Clinical trials are underway to explore if lurasidone may have any special benefits in those patients with marked cognitive symptoms in schizophrenia, due to its greater binding preference to 5 HT1A, 5 HT7 and α2C [28]. A trial to compare the efficacy of lurasidone over ziprasidone in ameliorating the cognitive symptoms of schizophrenia did show significant within-group improvement from baseline MCCB [Matrics Consensus Cognitive Battery] composite score (P < 0.026) and Schizophrenia Cognition Rating Scale (P < 0.001) in the lurasidone arm [35]. The drug is also being investigated for its potential benefit in schizophrenic patients with depressive symptoms following the evidence emerging from earlier studies that lurasidone did seem to improve the MADRS (Montgomery Asberg Depression Rating Scale) scores compared with placebo [36]. A study comparing lurasidone with risperidone in schizophrenia patients over 12 months showed that the improvement in PANSS score and CGI-S was comparable among the two drugs [37].
Safety & tolerability
Akathisia was reported as the most common adverse effect with as many as 15.1 % of patients in the lurasidone arm experiencing it. Akathisia (15 %), nausea (12 %), sedation (12 %), somnolence (11 %), parkinsonism (11 %), insomnia (8 %), agitation (6 %), anxiety (6 %), and dystonia (5 %) are some of the common reactions that would be expected during lurasidone therapy. Lurasidone did not show a significant increase in hyperglycemia, dyslipidemia, or weight gain. The drug does not appear to alter the QTc interval or cause any hemodynamic changes. Hyperprolactinemia may be observed during the early period of lurasidone therapy (3.6 %) versus placebo (0.6 %) [29, 34, 38–42].
Role of lurasidone in psychiatric practice
At the moment, there are no distinctive features that make lurasidone stand out among the atypical antipsychotics, although this may change if the drug does show a favourable response in improving cognitive and depressive symptoms of schizophrenia. The drug may be a better option in patients with comorbid conditions since its metabolic adverse effects are negligible. Nevertheless, there is a concern about akathisia, EPS and hyperprolactinemia, in which it resembles drugs such as risperidone and paliperidone. The results of trials evaluating lurasidone as monotherapy, add-on therapy and prophylaxis for bipolar depression are keenly awaited and may expand the spectrum for this novel antipsychotic.
Blonanserin
Blonanserin is an atypical antipsychotic approved in Japan and Korea that has a high affinity for dopamine D2, D3 and serotonin 5HT2A receptors [43, 44]. Unlike other atypical antipsychotics, its affinity for D2 receptors is much higher than that for 5HT2 receptors. A PET study done in schizophrenic patients with healthy controls showed that blonanserin had a striatal dopamine D2 receptor occupancy of 60.8 % at 8 mg, 73.4 % at 16 mg, and 79.7 % at 24 mg. The study helped to optimize the dose of blonanserin to a dose range of 12.9 to 22 mg/d for attaining a 70–80 % occupancy in the striatum [45]. The drug is dispensed at a dose between 4 and 24 mg/day in two divided doses. When adrenaline is given to a patient already on blonanserin it could precipitate a significant fall in blood pressure. The drug can also enhance the efficacy of antihypertensive drugs. CNS depressants such as barbiturates and alcohol are best avoided in a patient on blonanserin therapy as it could lead to enhanced depression of the central nervous system.
Pharmacokinetics
Being a drug that is extensively metabolized by cytochrome P450 enzyme CYP3A4, concomitant administration with other CYP3A4 inhibitors such as ketoconazole and protease inhibitors is contraindicated to avoid toxicity. As food increases the bioavailability of blonanserin, the dosing schedule of the patient should be adjusted accordingly [46]. The drug reaches maximal plasma concentration at 2 hours following dose administration. Although data on the drug pharmacokinetics in patients with hepatic and renal dysfunction is unavailable, the concentration of blonaserin is expected to raise in hepatic disease and so the drug should be used with caution in this population.[46]
Efficacy
The efficacy of blonanserin has been studied and proven in several randomized controlled clinical trials in comparison with placebo, haloperidol and risperidone in patients with schizophrenia [47, 48]. The drug showed a greater reduction than placebo in the PANSS total score after 6 weeks of therapy. Blonanserin also appeared to reduce the negative symptoms of schizophrenia, an effect which was not seen with haloperidol. These results were reproduced even when the BPRS score was used as the efficacy measure. Symptom severity was also found to be reduced in schizophrenia patients using the CGI-S scale, with blonanserin (1 week) showing an earlier response than haloperidol (3 weeks). Blonanserin has also been tested for its effect on cognitive function in twenty four first onset schizophrenia patients. Although the drug did show moderate improvement in cognitive symptoms, this effect has to be replicated in larger studies [49]. The drug also appears to show promise in the treatment of delirium in the ICU [50].
Safety & tolerability
The most common adverse reactions with blonanserin include Parkinson’s syndrome (35 %), akathisia (24.1 %), insomnia (22.4 %), hyperprolactinemia (19.6 %), dyskinesia (14 %), somnolence (11.8 %) and anxiety (11.2 %). Positive symptoms of schizophrenia such as excitability are also known to be worsened with blonanserin. Increased weight gain due to blonanserin is as common as that seen with haloperidol and risperidone.
Blonanserin is an antipsychotic that appears to show reasonable efficacy and tolerability in the acute and maintenance phases of schizophrenia. The drug has shown benefit in both positive and negative symptoms in schizophrenia. EPS and hyperprolactinemia are the most frequent problems observed with long term use of this drug. The drug has been most widely studied in Japan, Korea and recently marketed in India. A study is currently underway in China to study the efficacy and safety of this drug in Chinese subjects [51]. However more comparative studies are warranted especially with established front line atypical antipsychotics in larger populations before it becomes a routine choice in psychiatric practice (Table 3).
Cariprazine
Cariprazine, is a novel antipsychotic which is currently in the pipeline for the treatment of schizophrenia and bipolar disorder. It has a unique receptor binding profile with preferential affinity for the D3 and D2 receptors, and moderate affinity for the 5-HT1A receptor [52]. Located in limbic areas, ventral striatum and the thalamus, D3 dopamine receptors are shown to be involved in modulating memory function, speech, and attention in schizophrenia. Cariprazine is a novel antipsychotic drug candidate that exhibits partial agonism at D2/D3 receptors. The clinical effects of cariprazine are possibly related to the D3 and D2 receptors, with minimal 5-HT1A-related effects at therapeutic dose levels since the occupancy at 5HT1A receptor sites is only 18 %. These findings have been confirmed in a PET study performed in monkeys where cariprazine in increasing doses showed maximal occupancy of D2/D3 receptors. However the radioligands used in this study could not differentiate the subtype selectivity among dopamine receptors [53]. In another study that was carried out in male subjects with schizophrenia, D2 receptor occupancy was observed to be 69 % in the caudate nucleus and nucleus accumbens and 75 % in the putamen when cariprazine was given at a dose of 1.5 mg/d. Maximal occupancy of 90 % was seen only when the dose was increased to 3 mg/day [54]. Studies in animal models have shown that subnanomolar D3 antagonism coupled with nanomolar D2 activity results in favourable side effect profile with reduced extrapyramidal symptoms and also improved cognition [55] . The pro-cognitive effects of cariprazine have been attributed to its D3 agonism.
Cariprazine has completed phase III trials for the acute treatment of schizophrenia and is undergoing phase II trials for the treatment of bipolar disorder as an adjunct to antidepressants. Recently, Gedeon Richter labs announced the results of two phase III trials, the fixed and flexible dose studies. In the fixed dose study, total of 617 patients diagnosed of schizophrenia were given cariprazine in doses of 3 mg/day, 6 mg/day, aripiprazole 10 mg/day and placebo for a period of 6 weeks and additional 2 weeks safety follow up period when no drug was given. Cariprazine showed separation from placebo from the first week with dose of 6–9 mg/day and third week for the dose of 3–6 mg/day. The change in PANSS scores from the baseline was statistically significant compared to placebo [56]. Cariprazine was well tolerated with few adverse events. Most common side effects (>10 %) were akathisia, insomnia and headache. The drug which has been licensed to Forest Labs is currently awaiting FDA approval following an NDA submission in the far end of 2012.
Upcoming targets for schizophrenia
Glutamate receptors
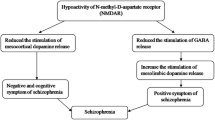
Glutamate is the predominant excitatory neurotransmitter in the brain. There is evidence implicating Group II metabotropic glutamate receptors (mGlu receptors) in modulating glutamatergic neurotransmission in the deranged synapses of the schizophrenic patient. NMDA receptor hypofunction with altered glutamate neurotransmission has been linked to the pathogenesis of schizophrenia. Among the glutamatergic drugs, LY2140023 has shown promising results in phase III trials. LY2140023 acts as an agonist at the metabotropic glutamate receptor 2/3(mGluR2/3). A proof of concept study showed that LY2140023 monohydrate was well tolerated and was associated with statistically significant changes in the PANSS scores when compared to placebo [57]. Another multicentric phase II study showed inconclusive results with, LY2140023 monohydrate not showing any distinct difference with olanzapine or placebo [58]. Currently four phase III trials are in progress for the treatment of schizophrenia. ADX 47273 is a positive allosteric modulator of mGlu5 receptor that has shown promising results in animal models of schizophrenia [59].
GABAergic signaling
Post-mortem studies from schizophrenic patients have revealed that abnormal GABAergic signaling is linked with poor cognitive function. The GABA-A receptors in the dorsolateral prefrontal cortex are the major mediators involved in memory. The level of GABA in these regions is controlled by the enzyme GAD67, an isoform of glutamic acid decarboxylase. Decreased expression of GAD67 would lead to reduced availability of GABA in the pyramidal neurons resulting in memory deficits. MK-0777 is an allosteric modulator of GABA-A receptors that increases neuronal synchronization in DLPFC (dorsolateral prefrontal cortex) and prevents the worsening of cognition. A phase II double blind randomized controlled trial investigating the efficacy of MK 0777 in improving cognitive symptoms in schizophrenia failed to show substantial benefit [60].
Cholinergic signaling
The cholinergic system is one of the pathways responsible for the cognitive deficits in schizophrenia. Neuronal nicotinic acetylcholine receptors (nAchR) are present in the anatomical structures (nucleus basalis of Meynert, medial septum, laterodorsal tegmental area and nucleus accumbens) implicated in the processes that are altered in schizophrenia such as attention, working memory and motivated behaviours. Nicotinic α7 receptors are found to be reduced in prefrontal cortex and hippocampal regions in patients with schizophrenia [61]. Interestingly, about 80 % of schizophrenia patients are smokers when compared to a smaller fraction in the general population reflecting a potential self-medication system. Several α7 receptor agonists are being evaluated for their usefulness in addressing the cognitive issues in schizophrenia.
AZD0328, an investigational α7 receptor agonist, was shown to improve the memory, cortical dopamine release, learning and attention processes in rodent models [62]. Recently a study carried out on nonhuman primates (rhesus macaques) showed that extremely low doses of a nicotinic α7 agonist can have profound acute and long-lasting beneficial consequences for improving cognition [63].
DMXB-A, another α7 agonist, was evaluated in a phase II trial with a randomised double blind, three arm (75 mg or 150 mg b.i.d or placebo) crossover study design. The primary end point was the improvement in MATRICS consensus cognitive battery tests. The study showed significant improvement in the clinical ratings of only negative symptoms (alogia and anhedonia) with improvements in working memory and attention being obscured by the practice effect at the end of study [64].
Tropisetron is a potent serotonin-3 (5-HT3) receptor antagonist widely used for the treatment of chemotherapy induced nausea and vomiting. Tropisetron also acts as a partial agonist at α7 nAChRs [65]. Sensory gating disturbances seen in schizophrenia which account for the cognitive deficits are manifested as deficient inhibition of P50 auditory evoked potential [66]. In particular, auditory sensory gating P50 deficits are correlated with neuropsychological deficits in attention, one of the principal cognitive disturbances in schizophrenia. A randomised, double blind, placebo controlled trial of tropisetron was performed to evaluate its efficacy in improving the cognition in schizophrenia. A total of 40 patients previously treated with risperidone(2–6 mg/day) were randomised to receive either tropisetron (10 mg/day) or placebo for a period of 8 weeks. Parameters measured were auditory sensory gating P50 deficits, Quality of Life Scale (QoLS), Cambridge Neuropsychological Test Automated Battery (CANTAB), and Positive and Negative Syndrome Scale (PANSS) scores. Tropisetron was well tolerated and significantly improved auditory sensory gating P50 deficits in non-smoking schizophrenia patients with no improvement in the placebo treated patients. The rapid visual information processing (sustained visual attention) task of CANTAB was significantly improved by tropisetron treatment when compared to placebo. However, PANSS scores remained unchanged with tropisetron [67, 68].
Varenicline, approved for smoking cessation, has also been tried as an adjunct for the treatment of cognitive deficits. It acts as a complete agonist at α7receptors and a partial agonist at α4β2 receptors. A recent study concluded that varenicline shows beneficial effects as an adjunctive treatment for the cognitive deficits of schizophrenia [69]. The drug showed statistically significant improvement in the digit symbol substitution test and the Wisconsin card sorting Test with no significant changes in the PANSS scores. In this study there were no reports of worsening depression or psychotic symptoms. Other studies done on varenicline have reported serious adverse events of worsening depression [67] and psychotic symptoms [70]. FDA has issued a black box warning mentioning that serious neuropsychiatric adverse events have been reported with the use of varenicline. Xanomeline, a M1/M4 agonist and M5 muscarinic receptor antagonist has also shown significant improvements in verbal memory and working memory. Besides this a number of other targets are being evaluated such as phosphodiesterase (PDE4 & PDE10), H3 receptors, neurokinin3, MAO-B and adenosine [71, 72]. Table 4 gives an overview of some of the molecules which are in early phase of drug development in schizophrenia.
Challenges in drug development of antipsychotics
The spate of failures among the new molecules for schizophrenia has led to many major pharmaceutical companies losing interest in the CNS drug discovery. Many pharmaceutical giants such as Novartis, Astra Zeneca, GlaxoSmithkline, Merck and Pfizer have pulled out of some of their drug discovery programmes and Novartis is currently focussing on the genetics of schizophrenia to identify novel drug targets [73]. The probability of success of a CNS drug to enter the market after successfully passing through the various phases of drug development is even lower than other research areas. Some of the major barriers to developing new successful antipsychotics include the lack of suitable animal models for the disease, poor construct validity of the disease, failure to understand the exact etiology of the disease, the absence of an appropriate biomarker that can correlate with disease progression and severity, avoidance of seriously mentally ill patients such as those with suicidal tendencies for ethical reasons in clinical trials of antipsychotics. Yet, with the rapid advances that are happening in neuroscience, it may not be a distant pipe dream to expect a blockbuster antipsychotic to hit the market within the next decade which is effective in relieving both negative and cognitive symptoms of schizophrenia.
Conclusion
Iloperidone, asenapine and lurasidone are the most recent atypical antipsychotics approved by the US FDA and EMA. The major advantage of iloperidone is the lesser chance of EPS. The sublingual formulation of asenapine maybe more acceptable to certain patients with swallowing difficulties, while the once daily lurasidone may be a convenient option for several patients with poor drug compliance. The adverse effect profile of these three antipsychotics is comparable to other second generation antipsychotics. Blonanserin is marketed in Japan, South Korea and India. Cariprazine is in the late stage of clinical development. Some of the novel targets that are in clinical development for schizophrenia include mGlu2/3 receptors, GABA receptors and nicotinic receptors. One hopes that these novel targets could be the turning point towards the surge of a new generation of molecules that would change the current nadir in antipsychotic drug development.
Abbreviations
- BPRS:
-
Brief Psychiatric Rating Scale
- CANTAB:
-
Cambridge Neuropsychological Test Automated Battery
- CGI-S:
-
Clinical Global Impression Severity scale
- DLPFC:
-
Dorsolateral prefrontal cortex
- EPS:
-
Extrapyramidal symptoms
- MATRICS:
-
Measurement and Treatment Research to Improve Cognition in Schizophrenia
- MCCB:
-
Matrics Consensus Cognitive Battery
- nAchR:
-
Neuronal nicotinic acetylcholine receptors
- PANSS-T:
-
Positive and negative syndrome scale total score
- PDE:
-
Phosphodiesterase
- QoLS:
-
Quality of Life Scale
References
Kasckow J, Felmet K, Zisook S (2011) Managing suicide risk in patients with schizophrenia. CNS Drugs 25:129–143
Valenstein M, Blow FC, Copeland LA et al (2004) Poor antipsychotic adherence among patients with schizophrenia: medication and patient factors. Schizophr Bull 30:255–264
Purcell SM, Wray NR, Stone JL et al (2009) Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460:748–752
Albers LJ, Musenga A, Raggi MA (2008) Iloperidone: a new benzisoxazole atypical antipsychotic drug. Is it novel enough to impact the crowded atypical antipsychotic market? Expert Opin Investig Drugs 17:61–75
McGuire P, Howes OD, Stone J, Fusar-Poli P (2008) Functional neuroimaging in schizophrenia: diagnosis and drug discovery. Trends Pharmacol Sci 29:91–98
Potkin SG. PET Findings with Iloperidone. 2008. Available from: http://www.nccmedical.com/images/portfolio%20samples/USPMHC_Poster_2011.pdf Accessed on Feb 18, 2013
Arif SA, Mitchell MM (2011) Iloperidone: a new drug for the treatment of schizophrenia. Am J Health Syst Pharm 68:301–308
Rado J, Janicak PG (2010) Iloperidone for schizophrenia. Expert Opin Pharmacother 11:2087–2093
Subramanian N, Kalkman HO (2002) Receptor profile of P88-8991 and P95-12113, metabolites of the novel antipsychotic iloperidone. Prog Neuropsychopharmacol Biol Psychiatry 26:553–560
Hiemke C, Baumann P, Bergemann N et al (2011) AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update. Pharmacopsychiatry 44:195–235
Potkin SG, Litman RE, Torres R, Wolfgang CD (2008) Efficacy of iloperidone in the treatment of schizophrenia: initial phase 3 studies. J Clin Psychopharmacol 28:S4–S11
Cutler AJ, Kalali AH, Weiden PJ, Hamilton J, Wolfgang CD (2008) Four-week, double-blind, placebo- and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol 28:S20–S28
Citrome L, Meng X, Hochfeld M, Stahl SM (2012) Efficacy of iloperidone in the short-term treatment of schizophrenia: a post hoc analysis of pooled patient data from four phase III, placebo- and active-controlled trials. Hum Psychopharmacol 27:24–32
Kane JM, Lauriello J, Laska E, Di Marino M, Wolfgang CD (2008) Long-term efficacy and safety of iloperidone: results from 3 clinical trials for the treatment of schizophrenia. J Clin Psychopharmacol 28:S29–S35
Weiden PJ, Cutler AJ, Polymeropoulos MH, Wolfgang CD (2008) Safety profile of iloperidone: a pooled analysis of 6-week acute-phase pivotal trials. J Clin Psychopharmacol 28:S12–S19
Vigneault P, Pilote S, Patoine D, Simard C, Drolet B (2012) Iloperidone fanapt(R), a novel atypical antipsychotic, is a potent HERG blocker and delays cardiac ventricular repolarization at clinically relevant concentration. Pharmacol Res. doi:10.1016/j.phrs.2012.03.008
Lavedan C, Licamele L, Volpi S et al (2009) Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol Psychiatry 14:804–819
Volpi S, Potkin SG, Malhotra AK, Licamele L, Lavedan C (2009) Applicability of a genetic signature for enhanced iloperidone efficacy in the treatment of schizophrenia. J Clin Psychiatry 70:801–809
Volpi S, Heaton C, Mack K et al (2009) Whole genome association study identifies polymorphisms associated with QT prolongation during iloperidone treatment of schizophrenia. Mol Psychiatry 14:1024–1031
Minassian A, Young JW (2010) Evaluation of the clinical efficacy of asenapine in schizophrenia. Expert Opin Pharmacother 11:2107–2115
Shahid M, Walker GB, Zorn SH, Wong EH (2009) Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol 23:65–73
Bishara D, Taylor D (2009) Asenapine monotherapy in the acute treatment of both schizophrenia and bipolar I disorder. Neuropsychiatr Dis Treat 5:483–490
FDA Psychopharmacologic Drugs Advisory Committee Meeting (2009) Available from http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PsychopharmacologicDrugsAdvisoryCommittee/UCM173876.pdf Accessed on Feb 18, 2013
Citrome L (2011) Role of sublingual asenapine in treatment of schizophrenia. Neuropsychiatr Dis Treat 7:325–339
Weber J, McCormack PL (2009) Asenapine. CNS Drugs 23:781–792
Kane JM, Cohen M, Zhao J, Alphs L, Panagides J (2010) Efficacy and safety of asenapine in a placebo- and haloperidol-controlled trial in patients with acute exacerbation of schizophrenia. J Clin Psychopharmacol 30:106–115
Szegedi A, Verweij P, Van Dujinhoven W (2010) Eficacy of asenapine for schizophrenia: comparison with placebo and comparative efficacy of all atypical antipsychotics using all available head-to-head randomized trials using meta-analytical techniques. Neuropsychopharmacology 35:S105
Ishibashi T, Horisawa T, Tokuda K et al (2010) Pharmacological profile of lurasidone, a novel antipsychotic agent with potent 5-hydroxytryptamine 7 (5-HT7) and 5-HT1A receptor activity. J Pharmacol Exp Ther 334:171–181
Owen RT (2011) Lurasidone: a new treatment option for schizophrenia. Drugs Today (Barc) 47:807–816
Samalin L, Garnier M, Llorca PM (2011) Clinical potential of lurasidone in the management of schizophrenia. Ther Clin Risk Manag 7:239–250
Nakazawa S, Yokoyama C, Nishimura N et al (2013) Evaluation of dopamine D(2)/D(3) and serotonin 5-HT(2)a receptor occupancy for a novel antipsychotic, lurasidone, in conscious common marmosets using small-animal positron emission tomography. Psychopharmacology (Berl) 225:329–339
Meyer JM, Loebel AD, Schweizer E (2009) Lurasidone: a new drug in development for schizophrenia. Expert Opin Investig Drugs 18:1715–1726
Citrome L (2011) Lurasidone for schizophrenia: a brief review of a new second-generation antipsychotic. Clin Schizophr Relat Psychoses 4:251–257
Citrome L (2011) Lurasidone for schizophrenia: a review of the efficacy and safety profile for this newly approved second-generation antipsychotic. Int J Clin Pract 65:189–210
Harvey PD, Ogasa M, Cucchiaro J, Loebel A, Keefe RS (2011) Performance and interview-based assessments of cognitive change in a randomized, double-blind comparison of lurasidone vs. Ziprasidone. Schizophr Res 127:188–194
Ogasa MLACJ (2009) Effect of lurasidone on depressive symptoms in patients with schizophrenia. Schizophr Bull 35:344–345
Citrome L, Cucchiaro J, Sarma K et al (2012) Long-term safety and tolerability of lurasidone in schizophrenia: a 12-month, double-blind, active-controlled study. Int Clin Psychopharmacol 27:165–176
Lurasidone (Latuda) for schizophrenia (2011) Med Lett Drugs Ther 53:13–14
Citrome L (2011) Iloperidone, asenapine, and lurasidone: a brief overview of 3 new second-generation antipsychotics. Postgrad Med 123:153–162
Cruz MP (2011) Lurasidone HCl (latuda), an oral, once-daily atypical antipsychotic agent for the treatment of patients with schizophrenia. P T 36:489–492
Nakamura M, Ogasa M, Guarino J et al (2009) Lurasidone in the treatment of acute schizophrenia: a double-blind, placebo-controlled trial. J Clin Psychiatry 70:829–836
Potkin SG, Ogasa M, Cucchiaro J, Loebel A (2011) Double-blind comparison of the safety and efficacy of lurasidone and ziprasidone in clinically stable outpatients with schizophrenia or schizoaffective disorder. Schizophr Res 132:101–107
Deeks ED, Keating GM (2010) Blonanserin: a review of its use in the management of schizophrenia. CNS Drugs 24:65–84
Ohno Y, Okano M, Imaki J, Tatara A, Okumura T, Shimizu S (2010) Atypical antipsychotic properties of blonanserin, a novel dopamine D2 and 5-HT2A antagonist. Pharmacol Biochem Behav 96:175–180
Tateno A, Arakawa R, Okumura, M et al (2013) Striatal and Extrastriatal Dopamine D2 Receptor Occupancy by a Novel Antipsychotic, Blonanserin: A PET Study With [11C]Raclopride and [11C]FLB 457 in Schizophrenia. J Clin Psychopharmacol
Saruwatari J, Yasui-Furukori N, Inoue Y, Kaneko S (2010) Effect of dose timing in relation to food intake on systemic exposure to blonanserin. Eur J Clin Pharmacol 66:899–902
Garcia E, Robert M, Peris F, Nakamura H, Sato N, Terazawa Y (2009) The efficacy and safety of blonanserin compared with haloperidol in acute-phase schizophrenia: a randomized, double-blind, placebo-controlled, multicentre study. CNS Drugs 23:615–625
Yang J, Bahk WM, Cho HS et al (2010) Efficacy and tolerability of Blonanserin in the patients with schizophrenia: a randomized, double-blind, risperidone-compared trial. Clin Neuropharmacol 33:169–175
Tenjin T, Miyamoto S, Miyake N et al (2012) Effect of blonanserin on cognitive function in antipsychotic-naive first-episode schizophrenia. Hum Psychopharmacol 27:90–100
Kato K, Yamada K, Maehara M et al (2011) Blonanserin in the treatment of delirium. Psychiatry Clin Neurosci 65:389–391
Efficiency Study to Investigate Blonanserin in Treatment of Schizophrenia When Compared With Risperidone. Clinical Trials.Gov [updated 19 January 2012, cited 2012 Apr 14]. Available from [http://clinicaltrials.gov/ct2/show/NCT01516424?term=blonanserin&rank=1]
Kiss B, Horvath A, Nemethy Z et al (2010) Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther 333:328–340
Seneca N, Finnema SJ, Laszlovszky I et al (2011) Occupancy of dopamine D2 and D3 and serotonin 5-HT1A receptors by the novel antipsychotic drug candidate, cariprazine (RGH-188), in monkey brain measured using positron emission tomography. Psychopharmacology (Berl) 218:579–587
Keator DB, Mukherjee J, Preda A (2009) Dopamine D2 and D3 receptor occupancy of cariprazine in schizophrenic patients. Schizophr Bull 35:154
Gyertyan I, Saghy K (2007) The selective dopamine D3 receptor antagonists, SB 277011-A and S 33084 block haloperidol-induced catalepsy in rats. Eur J Pharmacol 572:171–174
Citrome L. Cariprazine in Schizophrenia: Clinical Efficacy, Tolerability, and Place in Therapy. Adv Ther 2013 Jan 28
Patil ST, Zhang L, Martenyi F et al (2007) Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med 13:1102–1107
Kinon BJ, Zhang L, Millen BA et al (2011) A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J Clin Psychopharmacol 31:349–355
Liu F, Grauer S, Kelley C et al (2008) ADX47273 [S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1, 2, 4]-oxadiazol-5-yl]-piperidin-1- yl}-methanone]: a novel metabotropic glutamate receptor 5-selective positive allosteric modulator with preclinical antipsychotic-like and procognitive activities. J Pharmacol Exp Ther 327:827–839
Buchanan RW, Keefe RS, Lieberman JA et al (2011) A randomized clinical trial of MK-0777 for the treatment of cognitive impairments in people with schizophrenia. Biol Psychiatry 69:442–449
Ochoa EL, Lasalde-Dominicci J (2007) Cognitive deficits in schizophrenia: focus on neuronal nicotinic acetylcholine receptors and smoking. Cell Mol Neurobiol 27:609–639
Sydserff S, Sutton EJ, Song D et al (2009) Selective alpha7 nicotinic receptor activation by AZD0328 enhances cortical dopamine release and improves learning and attentional processes. Biochem Pharmacol 78:880–888
Castner SA, Smagin GN, Piser TM et al (2011) Immediate and sustained improvements in working memory after selective stimulation of alpha7 nicotinic acetylcholine receptors. Biol Psychiatry 69:12–18
Freedman R, Olincy A, Buchanan RW et al (2008) Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am J Psychiatry 165:1040–1047
Macor JE, Gurley D, Lanthorn T et al (2001) The 5-HT3 antagonist tropisetron (ICS 205-930) is a potent and selective alpha7 nicotinic receptor partial agonist. Bioorg Med Chem Lett 11:319–321
Cullum CM, Harris JG, Waldo MC et al (1993) Neurophysiological and neuropsychological evidence for attentional dysfunction in schizophrenia. Schizophr Res 10:131–141
Popkin MK (2008) Exacerbation of recurrent depression as a result of treatment with varenicline. Am J Psychiatry 165:774
Shiina A, Shirayama Y, Niitsu T et al (2010) A randomised, double-blind, placebo-controlled trial of tropisetron in patients with schizophrenia. Ann Gen Psychiatry 9:27
Shim JC, Jung DU, Jung SS et al (2012) Adjunctive varenicline treatment with antipsychotic medications for cognitive impairments in people with schizophrenia: a randomized double-blind placebo-controlled trial. Neuropsychopharmacology 37:660–668
Freedman R (2007) Exacerbation of schizophrenia by varenicline. Am J Psychiatry 164:1269
Tandon R, Nasrallah HA, Keshavan MS (2010) Schizophrenia, “just the facts” 5. Treatment and prevention. Past, present, and future. Schizophr Res 122:1–23
Ellenbroek BA (2012) Psychopharmacological treatment of schizophrenia: what do we have, and what could we get? Neuropharmacology 62:1371–1380
Abbott A (2011) Novartis to shut brain research facility. Nature 480:161–162
Lavedan C, Volpi S, Polymeropoulos MH, Wolfgang CD (2008) Effect of a ciliary neurotrophic factor polymorphism on schizophrenia symptom improvement in an iloperidone clinical trial. Pharmacogenomics 9:289–301
Food and Drug Administration. Drug approval package Fanapt (iloperidone) tablets: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/022192s007lbl.pdf; Accessed on Feb 18, 2013.
Food and Drug Administration. Drug approval package Saphris (asenapine) tablets: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022117s013lbl.pdf; Accessed on Feb 18, 2013.
Food and Drug Administration. Drug approval package Latuda (lurasidone hydrochloride) tablets: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/200603s009s013lbl.pdf; Accessed on Feb 18, 2013.
Wen YG, Shang DW, Xie HZ et al (2013) Population pharmacokinetics of blonanserin in Chinese healthy volunteers and the effect of the food intake. Hum Psychopharmacol. doi:10.1002/hup.2290
A Study of LY2140023 in Patients with Schizophrenia. Clinical Trials.gov [updated 30 March 2012, cited 2012 Apr 14] Available from [http://clinicaltrials.gov/ct2/show/NCT01307800?term=NCT01307800&rank=1]
A Comparison Study of LY2140023 and Aripiprazole in Schizophrenia Patients. Clinical Trials.gov [updated 17 February 2012, cited 2012 Apr 14] Available from [http://clinicaltrials.gov/ct2/show/NCT01328093?term=NCT01328093&rank=1]
Schlumberger C, Pietraszek M, Gravius A, Danysz W (2010) Effects of a positive allosteric modulator of mGluR5 ADX47273 on conditioned avoidance response and PCP-induced hyperlocomotion in the rat as models for schizophrenia. Pharmacol Biochem Behav 95:23–30
de Lucena D, Fernandes BS, Berk M et al (2009) Improvement of negative and positive symptoms in treatment-refractory schizophrenia: a double-blind, randomized, placebo-controlled trial with memantine as add-on therapy to clozapine. J Clin Psychiatry 70:1416–1423
Lieberman JA, Papadakis K, Csernansky J et al (2009) A randomized, placebo-controlled study of memantine as adjunctive treatment in patients with schizophrenia. Neuropsychopharmacology 34:1322–1329
Hashimoto K (2011) Glycine transporter-1: a new potential therapeutic target for schizophrenia. Curr Pharm Des 17:112–120
Wezenberg E, Verkes RJ, Ruigt GS, Hulstijn W, Sabbe BG (2007) Acute effects of the ampakine farampator on memory and information processing in healthy elderly volunteers. Neuropsychopharmacology 32:1272–1283
Evaluation of Single and Repeat Doses of GSK729327 in Healthy Volunteers. Clinical Trials.gov [updated 14 October 2010, cited 2012 Apr 14] Available from [http://clinicaltrials.gov/ct2/show/NCT00448890?term=NCT00448890&rank=1]
Jardemark K, Marcus MM, Malmerfelt A, Shahid M, Svensson TH (2012) Differential effects of AMPA receptor potentiators and glycine reuptake inhibitors on antipsychotic efficacy and prefrontal glutamatergic transmission. Psychopharmacology (Berl) 221:115–131
Gamma-Amino Butyric Acid (GABA)-A Alpha2/3 Study. Clinical Trials.gov [updated 14 October 2011, cited 2012 Apr 14]Available from [http://clinicaltrials.gov/ct2/show/NCT00129441?term=NCT+00129441&rank=1]
Treating Schizophrenia by Correcting Abnormal Brain Development. Clinical Trials.gov [updated 28 July 2011, cited 2012 Apr 14]Available from [http://clinicaltrials.gov/ct2/show/NCT00179465?term=NCT00179465&rank=1]
A Study to Examine the Pharmacodynamic Effects of GSK1034702 on Neurophysiological Biomarkers of Cognition in Nicotine Abstained Otherwise Healthy Smokers (MAA113746). Clinical Trials.gov [updated 9 June 2011, cited 2012 Apr 14]Available from [http://clinicaltrials.gov/ct2/show/NCT01371799?term=NCT01371799&rank=1]
Bradley SR, Lameh J, Ohrmund L et al (2010) AC-260584, an orally bioavailable M(1) muscarinic receptor allosteric agonist, improves cognitive performance in an animal model. Neuropharmacology 58:365–373
Pharmacologic and Clinical Testing of a D1 Agonist for Cognitive Enhancement in Neuropsychiatric Disorders. Clinical Trials.gov [updated 24 January 2012, cited 2012 Apr 14]Available from [http://clinicaltrials.gov/ct2/show/NCT01519557?term=NCT01519557&rank=1]
Arbabi M, Bagheri M, Rezaei F et al (2012) A placebo-controlled study of the modafinil added to risperidone in chronic schizophrenia. Psychopharmacology (Berl) 220:591–598
Efficacy, Safety, and Tolerability of SPD489 in Adults with Schizophrenia and Predominant Negative Symptoms. Clinical Trials.gov [updated 2 February 2012, cited 2012 Apr 14] Available from [http://clinicaltrials.gov/ct2/show/NCT00922272?term=NCT00922272&rank=1]
Clinical Trial of Tolcapone for Cognition in Schizophrenia. Clinical Trials.gov [updated 20 March 2012, cited 2012 Apr 14] Available from [http://clinicaltrials.gov/ct2/show/NCT00044083?term=NCT00044083&rank=1]
Meltzer HY, Massey BW, Horiguchi M (2012) Serotonin Receptors as Targets for Drugs Useful to Treat Psychosis and Cognitive Impairment in Schizophrenia. Curr Pharm Biotechnol. Jan 26.[Abstract]
Safety and Cognitive Function Study of EVP-6124 in Patients with Schizophrenia. Clinical Trials.gov [updated 5 March 2012, cited 2012 April 14] Available from [http://clinicaltrials.gov/ct2/show/NCT00968851?term=NCT00968851&rank=1]
McLean SL, Idris N, Grayson B et al (2011) PNU-120596, a positive allosteric modulator of alpha7 nicotinic acetylcholine receptors, reverses a sub-chronic phencyclidine-induced cognitive deficit in the attentional set-shifting task in female rats. J Psychopharmacol. doi:10.1177/0269881111431747
Efficacy, Safety, and Tolerability of TC-5619 as Augmentation Therapy to Improve Negative Symptoms and Cognition in Outpatients with Schizophrenia. Clinical Trials.gov [updated 6 March 2012, cited 2012 April 14] . Available from [http://clinicaltrials.gov/ct2/show/NCT01488929?term=NCT01488929&rank=1]
Roncarati R, Scali C, Comery TA et al (2009) Procognitive and neuroprotective activity of a novel alpha7 nicotinic acetylcholine receptor agonist for treatment of neurodegenerative and cognitive disorders. J Pharmacol Exp Ther 329:459–468
Marcus MM, Wiker C, Franberg O et al (2010) Adjunctive alpha2-adrenoceptor blockade enhances the antipsychotic-like effect of risperidone and facilitates cortical dopaminergic and glutamatergic, NMDA receptor-mediated transmission. Int J Neuropsychopharmacol 13:891–903
Halene TB, Siegel SJ (2008) Antipsychotic-like properties of phosphodiesterase 4 inhibitors: evaluation of 4-(3-butoxy-4-methoxybenzyl)-2-imidazolidinone (RO-20-1724) with auditory event-related potentials and prepulse inhibition of startle. J Pharmacol Exp Ther 326:230–239
Yang SW, Smotryski J, McElroy WT et al (2012) Discovery of orally active pyrazoloquinolines as potent PDE10 inhibitors for the management of schizophrenia. Bioorg Med Chem Lett 22:235–239
Southam E, Cilia J, Gartlon JE et al (2009) Preclinical investigations into the antipsychotic potential of the novel histamine H3 receptor antagonist GSK207040. Psychopharmacology (Berl) 201:483–494
Yoshikawa S, Hareyama N, Ikeda K et al (2009) Effects of TRK-820, a selective kappa opioid receptor agonist, on rat schizophrenia models. Eur J Pharmacol 606:102–108
Levkovitz Y, Mendlovich S, Riwkes S et al (2010) A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry 71:138–149
Muller N, Krause D, Dehning S et al (2010) Celecoxib treatment in an early stage of schizophrenia: results of a randomized, double-blind, placebo-controlled trial of celecoxib augmentation of amisulpride treatment. Schizophr Res 121:118–124
Pedersen CA, Gibson CM, Rau SW et al (2011) Intranasal oxytocin reduces psychotic symptoms and improves Theory of Mind and social perception in schizophrenia. Schizophr Res 132:50–53
Javitt DC, Buchanan RW, Keefe RS et al (2012) Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr Res 136:25–31
Study of Talnetant versus Placebo and Risperidone In Schizophrenia. Clinical Trials.gov [updated 1 March 2012, cited 2012 April 14] Available from [http://clinicaltrials.gov/ct2/show/NCT00103727?term=NCT00103727&rank=1]
Weiser M, Gershon AA, Rubinstein K et al (2012) A randomized controlled trial of allopurinol vs. placebo added on to antipsychotics in patients with schizophrenia or schizoaffective disorder. Schizophr Res 136:25–31
Rasagiline in the Treatment of Persistent Negative Symptoms of Schizophrenia. Clinical Trials.gov [updated 23 April 2011, cited 2012 April 14] Available from [http://clinicaltrials.gov/ct2/show/NCT00492336?term=NCT00492336&rank=1]
Bexarotene Augmentation of Antipsychotic Treatment for Chronic Schizophrenia. Clinical Trials.gov [updated 28 June 2010, cited 2012 April 14]. Available from [http://clinicaltrials.gov/ct2/show?term=NCT00141947&rank=2]
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
George, M., Amrutheshwar, R., Rajkumar, R.P. et al. Newer antipsychotics and upcoming molecules for schizophrenia. Eur J Clin Pharmacol 69, 1497–1509 (2013). https://doi.org/10.1007/s00228-013-1498-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-013-1498-4