
Abstract
Phylogeographical study of the brown macroalga, Sargassum aquifolium using nuclear internal transcribed spacer 2, plastidal RuBisCo spacer, and mitochondrial cytochrome oxidase subunit-III revealed the populations in Southeast Asia to be homogeneous. On the other hand, genetic differences were detected between populations from Southeast Asia and western Pacific Islands/Guam, suggesting the presence of genetic break between these regions. This further suggests that populations of S. aquifolium may have survived east of Sunda Shelf during the Last Glacial Maximum and recent recolonization led to homogeneity of the populations in the Sunda Shelf region. Recolonization could be facilitated by year-round reproduction of the populations and dispersal of germlings on floating thalli by coastal currents. Restricted current flow across Maluku Sea and directional equatorial current flows could have isolated the Pacific Island and Guam populations from those of Southeast Asia. Our results support the presence of multiple refugia as the source of different lineages of S. aquifolium populations with a lack of secondary contact in the post-glacial dispersal between Southeast Asia and western Pacific as the mechanisms behind the phylogeographical patterns observed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Southeast Asia is well known for having a complex geological history with fluctuating sea level during the glacial periods. The low sea level, up to 120 m below the present level in Pleistocene, resulted in formation of land bridges due to exposure of land masses in relatively shallow waters (Voris 2000). The largest land mass exposed, i.e., the Sunda Shelf, which included Malay Peninsula, Sumatra, Kalimantan, Java, Madura, Bali, and other smaller islands, created barriers for marine organisms (McManus 1985). Enclosed by Sunda Shelf and the surrounding land bridges were deeper basins, e.g., the South China Sea to the north, Sulu Sea in the northeast, and Celebes Sea and Flores Sea to the east. These basins served as refugia where isolation and differentiation of populations occurred (Barber 2000; Barber et al. 2006; Timm and Kochzius 2008). Post-glacial oceanographic conditions also contributed in shaping population structure of marine organisms. Monsoon currents promoted genetic connectivity of derived lineages (Wyrtki 1961). The strong downward Indonesian Throughflow (20 million m3/s), connecting Celebes Sea and Flores Sea through Makassar Strait (Fig. 1, Gordon and Fine 1996; Gordon 2005), has been identified as one of the most important directional current flows that contributed to high genetic connectivity among marine organisms in this region (Barber et al. 2006). On the other hand, this and other localized directional currents also serve as barriers blocking gene flows between populations (Barber et al. 2006). This complex geological history and the present oceanographic system are the key causes that contributed to Southeast Asia as one of the biodiversity hotspots and centers of marine endemism in the world (Hughes et al. 2002).

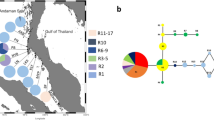
Haplotype distribution and haplotype network of Sargassum aquifolium for ITS2, Rbc spacer, and Cox3. Abbreviations of sample sites are given in Table 1. Pie chart size is proportional to sample size. Dominant currents are shown in solid lines (after Gordon and Fine 1996) and seasonally reversing currents in dashed lines (after Wyrtki 1961). Light gray area shows the coastal outline during Pleistocene maximum low sea level of −120 m (after Voris 2000). SEC South Equatorial Current; NEC North Equatorial Current; NECC North Equatorial Counter Current; NGCC New Guinea Coastal Current; ME Mindanao Eddy; HE Halmahera Eddy
Phylogeographical studies in Southeast Asia have revealed the populations of marine organisms to be highly structured. Genetic break between Sulawesi and Sunda Shelf was found in populations of false clown anemone fish (Timm and Kochzius 2008; Timm et al. 2012) and giant clam (Kochzius and Nuryanto 2008), with further genetic diversity contributed by subsequent recolonization. For stomatopods, genetic break was revealed between Papua New Guinea and Northern Indonesia where gene flow was restricted by limited current flow in Maluku Sea (Barber et al. 2006). These studies, however, mainly focused on the Coral Triangle region, and extensive sampling in Sunda Shelf region itself was only available for four species of seahorses (Lourie et al. 2005) wherein their population structures and genetic diversity were found to be related to the individual ecologies of these species. More study in the Sunda Shelf region itself is necessary to provide better understanding of its phylogeography and to lend weight to the suggestion of it as an independent unit for conversation management in Southeast Asia (Carpenter et al. 2011).
Most of the phylogeographical studies in Southeast Asia were on animals. Their lineage diversification and population connectivity among sites were highly dependent on the duration of their larval stages and direction of current flows (Crandall et al. 2008). Macroalgae, on the other hand, exhibited different mode of dispersal. Their immobile germlings with shorter dispersal distances (<km) than those of animal larvae may lead to higher differentiation between their populations. They could therefore provide a different perspective on understanding the forces shaping population structure of different marine organisms in the region. Extensive studies on macroalgae using multilocus molecular markers (Coyer et al. 2003; Provan et al. 2005; Olsen et al. 2010; Andreakis et al. 2007, 2009) have provided comprehensive information on the evolutionary history of marine organisms in North Atlantic. Recent phylogeographical studies using macroalgae (e.g., Cheang et al. 2010a) also revealed the importance of land bridges and oceanic current in contributing to the genetic diversification as well as mixing of genetic populations in northwestern Pacific.
The genus Sargassum (Phaeophyceae) is widely distributed in Southeast Asia. Members of this genus have a unique mode of dispersal. The dispersal distance of their germlings is very short, <1 m (Kendrick and Walker 1995), but they can also disperse for long distance in the form of drifting fronds with germlings attached (Ohno 1984). Southeast Asian region has a complex current system with a combination of monsoonal and directional current flows that may contribute to the dispersal, hence the mixing of populations of Sargassum spp.
Sargassum aquifolium (Turner) C. Agardh is a common brown macroalga growing on lower intertidal areas in many tropical reefs. It has been recorded in most Southeast Asian countries (Phillips 1995; Stiger et al. 2000; Ang et al. 2008). In this study, the nuclear internal transcribed spacer 2 (ITS2), plastidal RuBisCo spacer (Rbc spacer), and mitochondrial cytochrome oxidase subunit-III (Cox3) were used as molecular markers to investigate its phylogeographical pattern in Southeast Asia.
Materials and methods
Sample collection and DNA extraction
Samples were collected from 15 sampling sites within Southeast Asia by snorkeling and/or sampling during low tide (Fig. 1; Table 1). Young leaf tips were preserved in silica gel, and voucher plants were dried and stored as reference collections in the Simon F. S. Li Marine Science Laboratory Herbarium, Chinese University of Hong Kong. DNA was extracted following the methods described by Cheang et al. (2010a). Briefly, samples were crushed in liquid nitrogen, and genomic DNA was extracted by modified cetyltrimethylammonium bromide (CTAB) method. It was further purified by GENECLEAN II kit (Obiogene Inc.), following the manufacturer’s instructions.
Polymerase chain reaction and sequencing
The ITS2 and Cox3 were amplified with the following PCR profile: initial 2 min at 94 °C, 4 cycles of 30 s at 94 °C, 1 min at 47 °C and 1 min at 68 °C, 29 cycles of 25 s at 94 °C, 25 s at 55 °C, and 35 s at 72 °C. Rbc spacer was amplified with the following profile: initial 1 min at 94 °C, 40 cycles of 40 s at 94 °C, 30 s at 42 °C, and 45 s at 72 °C with final extension of 7 min at 72 °C. PCR was performed in a C1000™ thermal cycler (Bio-Rad Laboratory, Inc.). Primers for ITS2 and Rbc spacer were described in Mattio et al. (2008). Primers for Cox3 were newly designed (CoxF: GGGCCAGCATACTGTTGCGGT, CoxR: ACCAAGCGGCTGCTTCAAATCCA). Each 25 μL PCR contained 1.5 μL of DNA template, 2.5 μL of 10× buffer, 0.2 mM of each of dNTP, 0.2 μM of each primer, and 0.625 U of TaKaRa Ex Taq DNA polymerase (5 U/μL). PCR products were sequenced in both directions with the same primers by Macrogen (Macrogen Inc., Seoul, Korea). Sequences were aligned and edited together with the sequences of S. aquifolium available from GenBank by naked eyes using MEGA ver. 5 (Tamura et al. 2011).
Phylogeny and haplotype network construction
Samples of S. aquifolium were initially screened to confirm the identity before conducting subsequent analysis. Representative sequences from each haplotype of combined ITS2, Rbc spacer, and Cox3 sequences were initially screened to ensure they formed a monophyletic lineage with no misidentification. The sequences were aligned with those of other members of subgenus Sargassum spp. (Mattio et al. 2008, 2009) using Bayesian inference (BI) and maximum likelihood (ML) methods by MRBAYES 3.2.1 (Ronquist et al. 2012) and PhyML 3.0 (Guindon et al. 2010), respectively. Best-fit substitution model was determined by corrected Akaike information criterion (AICc) implemented in jMODELTEST 2.1.1 (Darriba et al. 2012). Turbinaria ornata (Turner) J. Ag., as suggested by Stiger et al. (2003) and Phillips et al. (2005), was used as outgroup. Any sequence that was not monophyletic was discarded to ensure consistency in subsequent analysis.
Phylogenetic relationship within S. aquifolium was investigated with ML using PhyML 3.0 and BI using MRBAYES 3.2.1. Best-fit substitution models were determined by jMODELTEST 2.1.1. Sargassum ilicifolium (Turner) C. Ag. was used as outgroup as it is closely related, being within the same subgenus Sargassum (Mattio et al. 2009). Significance of the branching was supported by 1,000 bootstrap in ML and posterior probability in BI analysis. For BI, two independent Markov Chain Monte Carlo (MCMC) searches were conducted until the divergence became small and stationary. Trees were sampled every 100 cycles in 1,500,000 generations, and first 25 % of trees were discarded as burn-in. Haplotype networks for the three markers were generated by TCS 1.21 (Clement et al. 2000).
Population structure analysis
Haplotype (h) and nucleotide (π) diversity (Nei 1987; Nei and Li 1989) were calculated in ARLEQUIN 3.5.1.2 (Excoffier and Lischer 2010). Tajima’s (1989) and Fu’s (1997) were calculated to test for the selective neutrality of the markers. Recent population bottleneck and population expansion can be indicated by negative Tajima’s D and Fu’s Fs. Pairwise Φ ST was calculated to estimate the level of gene flow between populations. Hierarchical analysis of molecular variance (AMOVA) with 10,000 permutations was conducted based on Φ ST to find out the best spatial groupings of populations. A Mantel test (Rousset 1997) with 10,000 permutations was used to test for the isolation by distance model by comparing the pairwise Φ ST and the matrix of geographical distances among sampling sites. Geographical distances were measured using the shortest distance between two sites by Google Earth (http://www.google.com/earth/index.html).
Demographic history
Demographic history of S. aquifolium was only conducted for Cox3 as no molecular clock is available for ITS2 and Rbc spacer. Mutation rate of Cox3 was calibrated to be 1.035 × 10−9–1.555 × 10−9 substitutions per site per year (Chan et al. 2013). The generation time of S. aquifolium was assumed to be 1 year as most Sargassum species have an annual cycle of regeneration (Ang 2006). Mismatch distribution was used to detect recent population expansion as categorized by unimodel distribution (Rogers and Harpending 1992) generated in ARLEQUIN 3.5.1.2. Time of expansion (in generation), t, can be calculated with τ = 2 μt, where τ is the crest of distribution calculated by ARLEQUIN and μ is the mutation rate of the marker per generation. A Bayesian skyline plot was used to estimate the demographic history of S. aquifolium. It was generated with BEAST 1.5.3 (Drummond et al. 2005) using MCMC sampling procedures with 108 steps in every 1,000 steps. Runs were repeated until effective sample size >200 in all parameters was reached, as recommended in the user manual (Drummond and Rambaut 2007). All runs were pooled in LogCombiner 1.7.3 with first 10 % of generations discarded as burn-in. Bayesian skyline plots were generated and visualized in Tracer 1.5.
Results
Genetic diversity
A total of 160, 115, and 151 sequences were obtained from 15 sampling sites and Genbank for ITS2 (297 bp), Rbc spacer (559 bp), and Cox3 (404 bp), respectively (Table 1; Fig. 1). Cox3 showed the highest variability (H = 0.782 ± 0.025, π = 0.1023 ± 0.0654). ITS2 and Rbc spacer showed similar variability (H = 0.230 ± 0.043, π = 0.0576 ± 0.0631 and H = 0.182 ± 0.049, π = 0.0370 ± 0.0381, respectively). A total of 8, 9, and 21 haplotypes were revealed for ITS2 (Genbank accession numbers for new sequences produced: KF826917–KF826920), Rbc spacer (KF826921–KF826927), and Cox3 (KF826928–KF826941), respectively, with 6, 11, and 18 polymorphic sites. No intragenomic variation was observed for all the markers as all the chromatographs showed clear and consistent peaks in both sequencing directions.
Interspecific and intraspecific phylogenetic relationship
HKY + G model was determined by jMODELTEST to be the best-fit substitution model to analyze the taxonomic status of S. aquifolium. Phylogenetic trees inferred from BI and ML with other members of the subgenus Sargassum showed all samples to be monophyletic (Fig. 2). All sequences were therefore used for subsequent analysis. HKY, GTR + G, and GTR + G models were determined by jMODELTEST to be the best-fit substitution model for ITS2, Rbc spacer, and Cox3, respectively. ML and BI analyses gave same tree topology and were supported by 1,000 bootstrap value and posterior probability (Figs. S1-S3, Electronic Supplementary Material).

Phylogenetic tree of representative sequences from each haplotype of combined ITS2, Rbc spacer, and Cox3 sequences from Sargassum aquifolium with other Sargassum spp. in subgenus Sargassum. Voucher numbers of sequences from Genbank are presented after species name. Representative sequences from each haplotype of S. aquifolium are labeled in black bar. Turbinaria ornata is used as outgroup. Posterior probabilities of Bayesian inference and bootstrap value of maximum likelihood are shown
Population structure
Tajima’s D gave significant negative value for ITS2 and Rbc spacer, but gave nonsignificant negative value for Cox3 (Table 1). Fu’s Fs gave significant negative value for all markers. Result of the two neutrality tests showed there has been population expansion in S. aquifolium. Most of the populations showed nonsignificant pairwise Φ ST values in ITS2 and Rbc spacer with shallow population structure (Tables S1–S2, Electronic Supplementary Material). Significant population differentiation was found in New Caledonia (NC) and Otres Beach, Cambodia (OB), and between these and other populations for ITS2 (pairwise Φ ST: 0.516–0.752 and 0.564–0.944, respectively, p < 0.05). For Rbc spacer, significant population differentiation was found in NC and Pantai Cenang, Malaysia (PC), and between these and other populations (pairwise Φ ST: 0.747–0.952 and 0.752–1.000, respectively, p < 0.05). In contrast, most pairwise Φ ST showed significant values for Cox3 (Table 2). High pairwise Φ ST values with other populations were found in Balibago, Calatagan, Philippines (BC, 0.502–1.000), Cocos Island (CC, 0.438–1.000), Fiji (FI, 0.423–0.960), Otres Beach (OB, 0.402–1.000), Pantai Cenang (PC, 0.453–1.000), Toguan Bay, Guam (TB, 0.583–0.980), and Vanuatu (VA, 0.433–1.000). Hierarchical AMOVA result is summarized in Table 3. For ITS2, highest interpopulation variation (%var = 51.08, Φ CT = 0.51) was found in the groupings: (1) Gulf of Thailand and South China Sea; (2) western Malaysian Peninsula and Java Sea; (3) Vanuatu, Solomon Islands and Fiji; (4) Guam; (5) NC. For Rbc spacer, highest interpopulation variation (%var = 58.46, Φ CT = 0.58) was found with the groupings: (1) Southeast Asia; (2) Vanuatu, Solomon Islands and Fiji; (3) Guam; (4) NC. For Cox3, shallower population structure was revealed with the same groupings as Rbc spacer (%var = 31.68, Φ CT = 0.32). Mantel test for ITS2, Rbc spacer, and Cox3 showed low regression coefficient (r = 0.1 × 10−5, p > 0.05; r = 2.6 × 10−5, p > 0.05; r = 0.5 × 10−5, p > 0.05) that did not fit into the isolation by distance model.
Demographic history
Demographic analysis was conducted in Cox3 only as no molecular clock is available for ITS2 and Rbc spacer. Mismatch distribution of Cox3 fits into the sudden expansion model (Fig. 3, sum of squared deviation 0.0059, p > 0.05). Estimated expansion time was 2.04–3.06 Mya, late Pliocene to early Pleistocene (τ = 2.559). Bayesian skyline plot showed gradual increase in population size since 0.25 Mya, late Pleistocene (Fig. 3).

Mismatch distribution (left) and Bayesian skyline plot (right) of Sargassum aquifolium from Cox3 sequences. Bar chart and line graph in mismatch distribution indicate the observed and expected frequency, respectively. Bayesian skyline plots are shown in effective population size with function of time (year before present in ka). 95 % confidence interval is shaded in gray
Discussion
Genetic variability
Overall, genetic variability found in S. aquifolium from Southeast Asia is comparable to that for other species of brown macroalgae. For example, 21 haplotypes of Cox3 were revealed in the present study, 17 in Ishige okamurae (Lee et al. 2012), and 9 in Undaria pinnatifida (Uwai et al. 2006) in northwestern Pacific. In contrast, genetic diversity among Sargassum spp. varies across regions and among species. For ITS2, eight haplotypes were revealed in the present study, two in S. hemiphyllum (Cheang et al. 2010a) in northwestern Pacific and 1 in S. muticum (Cheang et al. 2010b) from northwestern Pacific to Europe and North America. For Rbc spacer, nine haplotypes were revealed in the present study, three in S. hemiphyllum (Cheang et al. 2010a), one in S. muticum (Cheang et al. 2010b), and nine haplotypes in S. horneri (Hu et al. 2011). It was suggested that S. muticum evolved too recently to accumulate mutations causing the low genetic variability across a wide region (Bae et al. 2013). The genetic variability may thus reflect the difference in the evolutionary history experienced by these species across regions and/or the dispersal ability of the species that led to the present genetic pattern observed.
Among the three molecular markers used, Cox3 was found to have the highest variability in this study and was widely used in phylogeography of macroalgae in northwestern Pacific (Uwai et al. 2006; Hu et al. 2011; Lee et al. 2012). ITS2 and Rbc spacer have relatively lower variability than Cox3, but consistent genetic patterns could still be observed based on groupings of hierarchical AMOVA. They can serve as alternative markers besides mitochondrial genome for S. aquifolium, but markers with higher resolution power may be required for different species of Sargassum with different genetic variability.
Population structure
In general, lack of population structure was revealed within Southeast Asia, especially along the Sunda Shelf region, including the Gulf of Thailand. All populations within this region form a single group in hierarchical AMOVA based on Rbc spacer and Cox3 (Table 3). For ITS2, hierarchical AMOVA revealed one more group in Southeast Asia (i.e., South China Sea/Gulf of Thailand and West Peninsular Malaysia/West Java) which is mainly contributed by the differentiation of OB population from the other populations, as shown in pairwise differences (Table S1, Electronic Supplementary Material). However, shared haplotypes were observed between the two regions, suggesting that gene flow is not limited. More sampling sites would be needed to reveal any genetic break that may be present between South China Sea/Gulf of Thailand and Java Sea, as indicated in false clown anemonefish (Amphiprion ocellaris) and giant clam (Tridacna crocea) (Kochzius and Nuryanto 2008; Timm and Kochzius 2008).
The lack of population structure of S. aquifolium in Southeast Asia is unexpected given that complex geological history in the region is well established, involving fluctuating sea levels during Pleistocene that potentially restricted gene flow. Generalized concordant breaks found along the edge of Sunda Shelf on the Indian Ocean side and also along northern Java in the Java Sea support the presence of genetic barrier (i.e., the Sunda Shelf) (Carpenter et al. 2011). The incongruence in genetic pattern for S. aquifolium may be related to its rapid and successful dispersal after the Last Glacial Maximum so that no divergence of lineages could be revealed. Sargassum is capable of dispersing for long distance in the form of drifting fronds with germlings (Komatsu et al. 2007). Unlike other Sargassum spp. that exhibit only a single-peak reproductive season once a year (Ang 2006), S. aquifolium is capable of active reproduction throughout the whole year so that continuous production of germlings is possible and new recruits could be found every month (Yeong and Wong 2012). This reproductive strategy is in sharp contrast to animals with limited larval mobility (Carpenter et al. 2011) and allowed rapid re-establishment of gene flow with present-day monsoonal currents after the Glacial Maxima (Wyrtki 1961). While it is also possible that the variability of the markers used in this study may be too low to reveal any divergence of lineages, this is unlikely given that the same markers were used to reveal a high variability in the genetic lineage of S. ilicifolium, a closely related species (Chan et al. submitted).
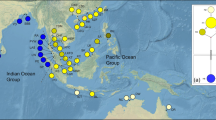
Hierarchical AMOVA showed differentiation in the spatial grouping of Guam (TB, PG, and CC), Pacific Islands (SO, VA, and FI), and NC, indicating that there may be genetic breaks elsewhere but not within the sampling area covered in this study. Samplings were carried out around southern Philippines, Sulawesi, and Java in Indonesia in the Coral Triangle region, but no populations of S. aquifolium were found. Further efforts need to be carried out to fill up this sampling gap between Southeast Asia and the Pacific Islands in order to confirm the presence of this genetic break. Nonetheless, private haplotypes were found in NC in all markers indicating isolation and limited gene flow in this site. Branches of south equatorial counter current (SECC) connect Fiji, Vanuatu, and Solomon Islands together and combine to form the New Guinea Coastal Current (NGCC) (Fig. 4, Benzie and Williams 1997; McGregor et al. 2008). NC is influenced by the downward branch of SECC so that upward gene flow may not be likely. In addition, NGCC flows along and east of Papua New Guinea to form the North Equatorial Counter Current (NECC). While this current circulation may allow genetic exchange between Pacific Islands, it may limit the gene flows to the west of Coral Triangle. Similarly, Guam populations were isolated from the west of Coral Triangle where private haplotypes were revealed. Such restricted current flow has also been suggested to limit genetic exchange in animal populations in the Coral Triangle region. In the study of three stomatopods, deep genetic divergence was revealed between Papua New Guinea and Indonesia and restricted current flow in the Maluku Sea in between was suggested to be a barrier that limited larval dispersal (Barber et al. 2006).

Detailed current flow among Pacific Islands with haplotype distribution of Cox3. Detailed current flows are modified from Benzie and Williams (1997) and McGregor et al. (2008). SEC South Equatorial Current; SECC South Equatorial Counter Current; NEC North Equatorial Current; NECC North Equatorial Counter Current; NGCC New Guinea Coastal Current; ME Mindanao Eddy; HE Halmahera Eddy
Demographic history
Estimated expansion time of S. aquifolium from mismatch distribution was 2.04–3.06 Mya, late Pliocene to early Pleistocene. During that period, sea level was fluctuating within 50–120 m below present level (Miller 2009). The scenario of S. aquifolium expansion and recolonization into the Sunda Shelf region resembles model III of contemporary phylogeographical pattern suggested by Maggs et al. (2008). The population structure of S. aquifolium observed today may be the result of populations deriving from two or more refugia between Southeast Asia and Pacific Islands/Guam. No secondary contact was achieved because of isolation by limited current exchange. It is hypothesized that S. aquifolium survived in east of Sunda Shelf during the Last Glacial Maximum and expanded to Guam and Pacific Islands. Recent recolonization to Sunda Shelf occurred, as indicated by gradual increase in population size in Bayesian skyline plot since 0.25 Mya, late Pleistocene (Fig. 3). Lack of population structure within the Sunda Shelf region may be the result of most recent recolonization. Previous population expansion cannot be revealed in Bayesian skyline plot (2.04–3.06 Mya from mismatch distribution analysis) as it is erased by the most recent signal in the Last Glacial Maximum (Grant et al. 2012). Therefore, the flat shape of Bayesian skyline plot before the Last Glacial Maximum cannot truly reflect the demographic history of this species.
Conclusion
Populations of S aquifolium were found to be genetically homogeneous within the Sunda Shelf region, but different from those from Guam and the western Pacific Islands. It is suggested that a genetic break likely exists in the middle of Coral Triangle where limited current flow through the Maluku Sea would have restricted gene flow between these regions. A scenario is proposed in which populations of S. aquifolium are suggested to persist east of Sunda Shelf and western Pacific during the Last Glacial Maximum and more recently recolonized in the Sunda Shelf, resulting in homogeneity of its population in the Sunda Shelf region due to recent expansion. Rapid expansion may be facilitated by year-round reproduction of this species, where germlings carried on floating thalli could be dispersed by complex oceanographic currents within this region.
References
Andreakis N, Procaccini G, Maggs C, Kooistra WHCF (2007) Phylogeography of the invasive seaweed Asparagopsis (Bonnemaisoniales, Rhodophyta) reveals cryptic diversity. Mol Ecol 16:2285–2299
Andreakis N, Kooistra WHCF, Procaccini G (2009) High genetic diversity and connectivity in the polyploid invasive seaweed Asparagopsis taxiformis (Bonnemaisoniales) in the Mediterranean, explored with microsatellite alleles and multilocus genotypes. Mol Ecol 18:212–226
Ang PO (2006) Phenology of Sargassum spp. in Tung Ping Chau Marine Park, Hong Kong SAR, China. J Appl Phycol 18:629–636
Ang PO, Leung SM, Choi MM, Cheang CC (2008) The genus Sargassum in the South China Sea region: a compilation list and preliminary biogeographical analysis. In: Phang SM, Lewmanomont K, Lim PE (eds) Taxonomy of Southeast Asia Seaweeds. Institute of Ocean and Earth Sciences, University of Malaya Monograph Series 2, pp 147–158
Bae DY, Ang PO, Boo SM (2013) Mitochondrial cox3 and trnW-I sequence diversity of Sargassum muticum. Aquat Bot 104:220–223
Barber PH (2000) A marine Wallace’s line? Nature 406:692–693
Barber PH, Erdmann MV, Palumbi SR (2006) Comparative phylogeography of three codistributed stomatopods: origins and timing of regional lineage diversification in the coral triangle. Evolution 60:1825–1839
Benzie JAH, Williams ST (1997) Genetic structure of giant clam (Tridacna maxima) populations in the West Pacific is not consistent with dispersal by present-day ocean currents. Evolution 51:768–783
Carpenter KE, Barber PH, Crandall ED et al (2011) Comparative phylogeography of the coral triangle and implications for marine management. J Mar Biol 2011:1–14
Chan SW, Cheang CC, Chirapart A et al (2013) Homogeneous population of the brown alga Sargassum polycystum in Southeast Asia: possible role of recent expansion and asexual propagation. PLoS ONE 8:e77662
Cheang CC, Chu KH, Ang PO (2010a) Phylogeography of the marine macroalga Sargassum hemiphyllum (Phaeophyceae, Heterokontophyta) in northwestern Pacific. Mol Ecol 19:2933–2948
Cheang CC, Chu KH, Fujita D et al (2010b) Low genetic variability of Sargassum muticum (Phaeophyceae) revealed by a global analysis of native and introduced populations. J Phycol 46:1063–1074
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Coyer JA, Peters AF, Stam WT, Olsen JL (2003) Post-ice age recolonization and differentiation of Fucus serratus L. (Phaeophyceae; Fucaceae) populations in Northern Europe. Mol Ecol 12:1817–1829
Crandall ED, Jones ME, Muñoz MM et al (2008) Comparative phylogeography of two seastars and their ectosymbionts within the coral triangle. Mol Ecol 17:5276–5290
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772
Drummond AJ, Rambaut A (2007) BEAST: bayesian evolutionary analysis by sampling trees. BMC Evolut Biol 7:214
Drummond AJ, Rambaut A, Shapiro B, Pybus OG (2005) Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22:1185–1192
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fu Y-X (1997) Statistical tests of neutrality against population growth, hitchhiking and background selection. Genetics 147:915–925
Gordon AL (2005) Oceanography of the Indonesian seas and their throughflow. Oceanography 18:14–27
Gordon AL, Fine RA (1996) Pathways of water between the Pacific and Indian oceans in the Indonesian seas. Nature 379:146–149
Grant WS, Liu M, Gao T, Yanagimoto T (2012) Limits of Bayesian skyline plot analysis of mtDNA sequences to infer historical demographies in Pacific herring (and other species). Mol Phylogenet Evol 65:203–212
Guindon S, Dufayard J-F, Lefort V et al (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Hu Z-M, Uwai S, Yu S-H et al (2011) Phylogeographic heterogeneity of the brown macroalga Sargassum horneri (Fucaceae) in the northwestern Pacific in relation to late Pleistocene glaciation and tectonic configurations. Mol Ecol 20:3894–3909
Hughes TP, Bellwood DR, Connolly SR (2002) Biodiversity hotspots, centres of endemicity, and the conservation of coral reefs. Ecol Lett 5:775–784
Kendrick GA, Walker DI (1995) Dispersal of propagules of Sargassum spp. (Sargassaceae: Phaeophyta): Observations of local patterns of dispersal and consequences for recruitment and population structure. J Exp Mar Biol Ecol 192:273–288
Kochzius M, Nuryanto A (2008) Strong genetic population structure in the boring giant clam, Tridacna crocea, across the Indo-Malay Archipelago: implications related to evolutionary processes and connectivity. Mol Ecol 17:3775–3787
Komatsu T, Tatsukawa K, Filippi JB et al (2007) Distribution of drifting seaweeds in eastern East China Sea. J Mar Syst 67:245–252
Lee KM, Yang EC, Coyer JA et al (2012) Phylogeography of the seaweed Ishige okamurae (Phaeophyceae): evidence for glacial refugia in the northwest Pacific region. Mar Biol 159:1021–1028
Lourie SA, Green DM, Vincent ACJ (2005) Dispersal, habitat differences, and comparative phylogeography of Southeast Asian seahorses (Syngnathidae: Hippocampus). Mol Ecol 14:1073–1094
Maggs CA, Castilho R, Foltz D et al (2008) Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology 89:S108–S122
Mattio L, Payri CE, Stiger-Pouvreau V (2008) Taxonomic revision of Sargassum (Fucales, Phaeophyceae) from French Polynesia based on morphological and molecular analyses. J Phycol 44:1541–1555
Mattio L, Payri CE, Verlaque M (2009) Taxonomic revision and geographic distribution of the subgenus Sargassum (Fucales, Phaeophyceae) in the Western and central Pacific Islands based on morphological and molecular analyses. J Phycol 45:1213–1227
McGregor HV, Gagan MK, McCulloch MT et al (2008) Mid-Holocene variability in the marine 14C reservoir age for northern coastal Papua New Guinea. Quat Geochronol 3:213–225
McManus JW (1985) Marine speciation, tectonics, and sea level changes in Southeast Asia. In: Proceedings of the 5th International Coral Reef Congress, Tahiti 4: 133–138
Miller KG (2009) Sea level change, last 250 million years. http://www.springerreference.com/docs/html/chapterdbid/77470.html. Accessed 19 July 2007
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nei M, Li J (1989) Variances of the average numbers of nucleotide substitutions within and between populations. Mol Biol Evol 6:290–300
Ohno M (1984) Observation on the floating seaweed on the near-shore waters of the southern Japan. Hydrobiologia 116⁄117: 408–412
Olsen JL, Zechman FW, Hoarau G et al (2010) The phylogeographic architecture of the fucoid seaweed Ascophyllum nodosum: an intertidal “marine tree” and survivor of more than one glacial-interglacial cycle. J Biogeogr 37:842–856
Phillips N (1995) Biogeography of Sargassum (Phaeophyta) in the Pacific basin. In: Abbott IA (ed) Taxonomy of economic seaweeds with reference to some Pacific Species, vol 5. California Sea Grant College Program, La Jolla, pp 107–145
Phillips NE, Smith CM, Morden CW (2005) Testing systematic concepts of Sargassum (Fucales, Phaeophyceae) using portions of the rbcLS operon. Phycoll Res 53:1–10
Provan J, Wattier RA, Maggs CA (2005) Phylogeographic analysis of the red seaweed Palmaria palmata reveals a Pleistocene marine glacial refugium in the English Channel. Mol Ecol 14:793–803
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Ronquist F, Teslenko M, van der Mark P et al (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Stiger V, Horiguchi T, Yoshida T et al (2000) Phylogenetic relationships of Sargassum (Sargassaceae, Phaeophyceae) with reference to a taxonomic revision of the section Phyllocystae based on ITS-2 nrDNA sequences. Phycol Res 48:251–260
Stiger V, Horiguchi T, Yoshida T et al (2003) Phylogenetic relationships within the genus Sargassum (Fucales, Phaeophyceae), inferred from ITS-2 nrDNA, with an emphasis on the taxonomic subdivision of the genus. Phycol Res 53:1–10
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Peterson D, Peterson N et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evolut 28:2731–2739
Timm J, Kochzius M (2008) Geological history and oceanography of the Indo-Malay Archipelago shape the genetic population structure in the false clown anemonefish (Amphiprion ocellaris). Mol Ecol 17:3999–4014
Timm J, Planes S, Kochzius M (2012) High similarity of genetic population structure in the false clown anemonefish (Amphiprion ocellaris) found in microsatellite and mitochondrial control region analysis. Conserv Genet 13:693–706
Uwai S, Yotsukura N, Serisawa Y et al (2006) Intraspecific genetic diversity of Undaria pinnatifida in Japan, based on the mitochondrial cox3 Gene and the ITS1 of nrDNA. Hydrobiologia 553:345–356
Voris HK (2000) Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. J Biogeogr 27:1153–1167
Wyrtki K (1961) Physical oceanography of the Southeast Asian waters. NAGA Report Vol. 2, The University of California, Scripps Institution of Oceanography, La Jolla, California
Yeong M-LB, Wong C-L (2012) Seasonal growth rate of Sargassum species at Teluk Kemang, Port Dickson, Malaysia. J Appl Phycol 25:805–814
Acknowledgments
We thank Tharith C (Cambodia), Phang Siew Moi (Malaysia), Edna Ganzon-Fortes (Philippines), Jeff Low (Singapore), Jaruwan Mayakun, and Achana Prathep (Thailand), for their hospitality during our collecting trips and for help with the collection of samples. Tsang Ho Leung assisted in the collection in Guam. This study was supported by RGC-CRF Project 460108 to P Ang.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Uthicke.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chan, S.W., Cheang, C.C., Yeung, C.W. et al. Recent expansion led to the lack of genetic structure of Sargassum aquifolium populations in Southeast Asia. Mar Biol 161, 785–795 (2014). https://doi.org/10.1007/s00227-013-2377-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00227-013-2377-3