
Abstract
Despite considerable interest in physiology, evolution and life history of Antarctic marine invertebrates, only a limited number of studies have examined the genetic variability and diversity patterns of these organisms. Moreover, understanding and characterizing patterns of Antarctic biodiversity has taken on a degree of urgency because of potential impacts of global warming. To expand an understanding of the evolutionary history of Antarctic marine invertebrates, the genetic diversity of the crinoid Promachocrinus kerguelensis Carpenter, 1888 was investigated, which is documented to have a circumpolar distribution extending to subantarctic islands. Specimens of P. kerguelensis were collected from the western side of the Antarctic Peninsula, and the subantarctic islands South Georgia, South Sandwich and Bouvetøya Island from 2001 to 2004. P. kerguelensis was previously subject to morphological review that confirmed the taxonomic recognition of only one species. The wide distribution and reported high dispersal capability for P. kerguelensis predicts one large panmictic population. In contrast, nucleotide sequence data from mitochondrial cytochrome oxidase subunit I and cytochrome b genes, collected herein, reveal distinct genetic structure and cryptic speciation within P. kerguelensis. In the Antarctic Atlantic sector alone, there were at least five “species-level” clades. Some of these clades are geographically limited, and most exist in sympatry. The largest and most widespread of these clades was examined to help elucidate connectivity along the subantarctic islands of the Scotia Arc and the Antarctic Peninsula. Within this clade, most genetic diversity was contained within populations, but significant differences were present between regions (Antarctic Peninsula, South Sandwich Is., South Georgia, Bouvetøya Is.), suggesting a corresponding lack of gene flow. Given that P. “kerguelensis” is a well-studied taxon, the finding of considerable genetic diversity within the Atlantic sector alone suggests that the recognized diversity of Antarctica’s benthic marine life may be underestimated, and will rise dramatically with phylogeographic analyses of putative widespread species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antarctic waters are often thought of as containing an endemic, cold-adapted fauna with many species exhibiting a circumpolar distribution (e.g. Hedgpeth 1969; Dayton 1990; Dell 1990; Sará et al. 1992). Many taxa (e.g. decapod crustaceans, teleost fish) have reduced diversity (Clark 1990), whereas others have been opportunistic and undergone radiations (e.g. notothenoid fish, Eastman and Clarke 1998; serolid crustaceans, Held 2000). One of the main explanations for these patterns is the cooling and thermal isolation of Antarctica likely caused by the separation of Antarctic and South American continents, and the concomitant establishment of the Antarctic circumpolar current (ACC) approximately 28–41 Ma (e.g. Lawver and Gahagan 2003; Scher and Martin 2006). Although there has been considerable work on the biology of Antarctic marine fauna and estimates of Antarctic biodiversity in general (e.g. Arntz et al. 1997; Arntz and Rios 1999; Gray 2001; Gutt et al. 2004), few studies have focused on understanding genetic patterns, speciation processes, and/or environmental mechanisms that have lead to recent Antarctic biodiversity (but see Allcock et al. 1997; Chen et al. 1997; Page and Linse 2002; Shaw et al. 2004; Held and Wägele 2005). Thus, commonly accepted taxonomic hypotheses based on morphology, hypotheses of faunal distribution (e.g. circumpolarity, endemism) and the influence of processes shaping evolutionary history (e.g. ACC formation) are largely untested (see Thatje et al. 2005).
Testing these hypotheses is important for understanding the degree to which information can be extrapolated from apparently “conspecific” animals across different Antarctic regions, for example from the Ross Sea and Weddell Sea which are on opposite sides of the continent. More urgently, parts of Antarctica, including the Peninsula, are known to be warming at several times the rate of the global average (Vaughan et al. 2003) and consequently its biodiversity is under great threat. If many of the widely distributed Antarctic taxa represent cryptic species complexes, such as those more usually recognised from lower latitudes (Palumbi 1992; Knowlton 1993, 2000), then biodiversity estimates for Antarctica may be vastly underestimated, and species may disappear before they are described.
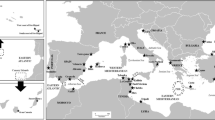
To begin to address such issues, we explored evolutionary patterns of genetic structure in a widespread, circumpolar endemic invertebrate, the comatulid crinoid Promachocrinus kerguelensis (Family Antedonidae). This is one of the largest, most abundant and most widely distributed crinoids on the Antarctic shelf and slope, ranging from 20 to 2,100 m in depth (Speel and Dearborn 1983); (Fig. 1). P. kerguelensis is highly fecund with buoyant eggs that are assumed to be capable of dispersing with surface currents; estimates indicate larval duration could be as long as 3 months (McClintock and Pearse 1987) although this is incongruent with data for other comatulid crinoids. Adult P. kerguelensis are also powerful swimmers (Marr 1963), and are prone to rafting processes as they perch on sponges, thus allowing additional post-larval dispersal. Although known to be morphologically variable, thorough taxonomic revision recognized only a single species in the genus (John 1938). A later monograph (Clark and Clark 1967) synthesized all previous known descriptions and records, encompassing samples from the type locality of Îles Kerguelen, east Antarctica, the Antarctic Peninsula and South Shetland Islands, the Ross and Davis Seas, and the subantarctic islands of South Georgia, Bouvetøya and the South Sandwich Islands. They also upheld the hypothesis of a single species, and later work by Speel and Dearborn (1983) concurred after inspecting 1,451 specimens from various localities.

Distribution of Promachocrinus “kerguelensis”. Dots represent published records (Marr 1963; Speel and Dearborn 1983) and stars represent regions sampled in this study. The path of the Scotia Arc is shown as a dashed line. The dark grey area shows oceanic waters usually south of the polar front. Base map supplied by the Census of Antarctic Marine Life, courtesy of the Australian Antarctic Division
Here we investigate the genetic diversity in P. kerguelensis using mitochondrial cytochrome oxidase I and cytochrome b sequence data. If P. kerguelensis was a single species that maintains a continuous circumpolar distribution around Antarctica, we would expect panmixia among regions, or at least gene flow between adjacent regions. Because of logistical problems in accessing adequate samples from the entire coastline of the Antarctic continent, we sampled extensively within the Atlantic sector, including subantarctic islands. Employing standard phylogeographic methods, we examined patterns of genetic diversity and isolation in P. kerguelensis to explore phylogeographic structure as it related to hypotheses such as panmixia, circumpolarity, and morphological species boundaries.
Materials and methods
Sampling
Specimens of P. kerguelensis Carpenter, 1888 were collected from the western side of the Antarctic Peninsula (Fig. 1) using the R/V Laurence M. Gould (December 2001 and 2004) equipped with a Blake trawl, Smith-McIntyre grab, wire dredge or epibenthic sled. The depth of sampled sites ranged from 116 to 1,170 m (S1). Selected vouchers were photographed, and all samples were frozen or fixed in ethanol. Specimens from South Georgia, South Sandwich Islands and Bouvetøya Island (Fig. 1) were collected during the ICEFISH 2004 cruise on the R/V Nathaniel Palmer with Blake or otter trawls, and fixed in ethanol. Collection details, sample data and Genbank accessions are listed in S1. Voucher specimens have been deposited at the Smithsonian Museum of Natural History. Individuals used in analyses were representative of the size range (approximately 2–20 cm arm length) observed in the wild. Selected voucher photographs are available on http://gump.auburn.edu/halanych/iic/index.html.
DNA extraction, PCR and sequencing
Tissue samples were extracted using a Qiagen Dneasy® Tissue Kit according to the manufacturer’s instructions. A 623-bp fragment of cytochrome oxidase I (COI) was amplified using the Folmer et al. (1994) primers and the following cycling conditions: initial denaturation 95°C, 2 min; 35 cycles of denaturation 94°C, 30 s, annealing 44–45°C, 1 min; extension 68°C, 1 min; final extension 68°C, 7 min. A 663-bp fragment of cytochrome b (CytB) was amplified using the primers StCytbF and StCytbR (G. Rouse et al. personal communication) with conditions: initial denaturation 94°C, 2 min; 35 cycles of denaturation 94°C, 30 s, annealing 43°C, 30 s, and extension, 68°C, 1 min; final extension 68°C, 7 min. Purified PCR products were bi-directionally sequenced on a Beckman CEQ 8000. As there were no indels or deletions, the sequences were easily aligned manually in Se–Al v2.0a11 (Rambaut 2002) and the alignment (including tree topologies) deposited to TreeBASE (S1780). Using McClade v4.06 (Maddison and Maddison 2003), inferred protein sequences aided alignment and were used to check for sequencing errors.
Analyses
Two additional Antedonidae species (Florometra serratissima AF049132 and Heliometra glacialis maxima SAMK2145), served as outgroups. These two genera, together with Promachocrinus, are placed in the subfamily Heliometrinae (Clark and Clark 1967). A partition homogeneity test in PAUP* (1,000 replicates, heuristic searches limited to 100,000 rearrangements, TBR branch swapping and ten random sequence additions) indicated no conflicting signal (P = 0.177), so CytB and COI sequence data were combined.
TCS 1.21 (Clement et al. 2000) was used to generate a haplotype network representing intraspecific relationships not dependent on hierarchical evolutionary processes. However, the 95% plausible set of linkages resulted in multiple unconnected networks (Fig. 2), thus a phylogenetic approach was also necessary. We used only unique haplotypes for maximum parsimony (MP) in PAUP* (Swofford 2002) and likelihood estimations in MRBAYES v3.1.2 (Huelsenbeck and Ronquist 2001) for phylogenetic analyses. MP analyses employed heuristic searches with 100 random additions and TBR branch swapping. Bootstrapping (10,000 iterations) was carried out on unique haplotypes differing by more than one mutational step. For Bayesian analyses, the data were set as two unlinked partitions, with models selected via the AIC in MrModeltest v2.2 (Nylander 2002) (COI—HKY + I + G; CytB—GTR + I + Γ). We used two sets of four simultaneous chains (1 cold) run to 106 generations. The first 4,000 generations were discarded as burn-in and remaining trees were sampled every 200 generations. Average corrected pairwise distances within and between clades were calculated in PAUP*, implementing the model GTR + I + Γ selected using MODELTESTv3.7 (Posada and Crandall 1998). A generalised molecular clock for echinoderms was used to crudely date cladogenic events (3.1–3.5%/MY—Lessios et al. 1999; McCartney et al. 2000).

Haplotype networks of P. kerguelensis produced by TCS1.21 (Clement et al. 2000) (95% set). Empty circles represent unsampled, or possibly extinct haplotypes. Networks are labelled with clade haplotype numbers. Numbers outside of shapes show frequency of haplotype occurrence if greater than one
Haplotypic and nucleotide diversity (Nei 1987) in the largest clade (A; n = 75), were calculated using ARLEQUIN v2.0 (Schneider et al. 2000). Additionally, we conducted a hierarchical AMOVA, and regional pairwise FST comparisons based on haplotypic frequencies, assessing significance with permutation tests (1,000 iterations). Model-based estimates of gene flow, ϕST (Tamura and Nei 1993, with gamma 0.4908 from MODELTEST) were also calculated. We tested the frequency distribution of pairwise differences between haplotypes in the sample against a model of rapid expansion growth (mismatch analysis) (Rogers and Harpending 1992) using parametric bootstrapping with 10,000 replicates (Schneider and Excoffier 1999).
Results
Sequence diversity, networks and phylogenetic structure
Sequences from 124 individuals of P. kerguelensis consisted of 54 unique mitochondrial haplotypes. Of 1,286 aligned positions (COI—623 bp, CytB—663 bp), 216 positions were variable (COI—90 bp, CytB—126 bp) and 185 were parsimony informative (COI—78 bp, CytB—107 bp). Individually, TCS results for COI and CytB resulted in five congruent haplotype networks. However, when data were combined, six networks were produced (herein named A–F, see Fig. 2); a long divergence within one network was pushed outside the 95% limits of parsimony reconnection, forming two networks (B + C). Variation within a clade (0–0.39%) was always less than 1/10 the amount among clades (4.18–14.72%), the exception being the low variation between clades B and C (1.88%; Table 1).
Examining evolutionary relationships among all clades, Bayesian and parsimony produced consistent topologies, and nodes corresponding to haplotype networks were highly supported (bootstrap values >90; posterior probabilities >0.90) as monophyletic in both (see Fig. 3). The Bayesian analyses showed an unresolved trichotomy of clades A + B + C + D being sister to E + F. MP resolved A as the sister to B + C, but only by a single, weakly supported node.

Bayesian consensus tree for P. kerguelensis generated in MrBayes v3.1.2 (Huelsenbeck and Ronquist 2001). Number in parentheses show the number of individuals of observed for a given haplotype if greater than one. Posterior probabilities above, maximum parsimony (MP) bootstraps below. Models implemented from MrModeltest v2.2 (Nylander 2002) were HKY + I + G for COI and GTR + I + Γ for CytB. Note that MP recovered a clade A + B + C with bootstrap support of 67
Geographic and depth distribution of clades
All clades occurred in multiple localities. At the regional scale, the southwestern Antarctic Peninsula (below Bransfield Strait) contained C and F, and the north contained clades A, B, C, D and E (see Fig. 4). Clades A and E were found in South Georgia; clades A and C in South Sandwich Is., but Bouvetøya Is. contained only clade A. At the local scale, all sites with more than two individuals hosted multiple clades; Bouvetøya Is. being the exception, with all five sites containing only clade A (n = 37). Clade D was only found at three sites closest to Deception Island. The single haplotype of Clade E occurred at two sites in the Bransfield Straight (Antarctic Peninsula), and in South Georgia.

Promachocrinus clade frequency and distribution. Sites in subantarctic regions have been pooled. Small circle represents 1–5 individuals; medium circle 6–10, large circle >11. Actual sample size next to circles
Most clades were found at a variety of depths. Clade A was found from 116 to 315 m; Clade B from 203 to 277 m; Clade C from 185 to 1,170 m; Clade D from 203 to 1,170 m; Clade E from 185 to 204 m; and Clade F from 205 to 368 m. Depth comparisons must remain preliminary as only one site deeper than 370 m was sampled, but ranges for clades overlapped and showed no evidence for depth-related isolation.
Population diversity and demography in clade A
The regionally isolated population structure revealed here for clade A (the largest network in Fig. 2), is discordant with panmixia. Clade A haplotypic diversity calculated in ARLEQUIN was high within sites (mean h = 0.797) and nucleotide diversity was low (mean π = 0.0025). AMOVA found highly significant genetic subdivision among sites and among regions (Table 2). There was no significant subdivision within regions, so we pooled samples within these for pairwise regional FST and ϕST analyses (Table 3). The FST comparisons indicated a lack of gene flow between the Antarctic Peninsula, South Sandwich Is., and Bouvetøya Is., but not South Georgia (which was expected with a low sample size). However, ϕST detected significant subdivision between all regions except the Antarctic Peninsula and South Georgia. The assumed ancestral haplotype indicated by TCS was found at sites along the Antarctic Peninsula (n = 1), South Sandwich Is. (n = 2), and Bouvetøya Is. (n = 5). Mismatch analysis on pooled South Sandwich Is., Bouvetøya Is. and Antarctic Peninsula samples could not reject the sudden expansion model of population growth. The variance was too small to make calculations for South Georgia.
Discussion
Promachocrinus kerguelensis consists of a complex of related species. Mitochondrial data revealed six distinct, reciprocally monophyletic clades, which likely represent at least five species. The percent divergence between clades is highly congruent with species-level distinctions in other echinoderms (Lessios et al. 2001; Waters et al. 2004; Helgen and Rouse 2006). We show that cryptic species are present in geographic and bathymetric sympatry. Given that our sampling focused on the Atlantic sector, we also expect that the full diversity of Promachocrinus is much higher.
Taxonomic revision of P. kerguelensis recognises a single species, but high levels of external morphological variation have been acknowledged. The previously recognized P. vanhöffenianus Minckert differed by details of cirri and arm segment shape, and most distinctively, having covering plates along most ambulacral grooves, but these features were viewed as correlated and indicative of immaturity (Clark and Clark 1967). Similarly, P. joubini Vaney also showed juvenile characteristics, and was considered conspecific with P. kerguelensis by Clark and Clark (1967). Speel and Dearborn (1983) noted that there were no geographical correlations with morphological variation in food groove patterns in P. kerguelensis, and concluded that the range of variation present was as great within as among locations. Given our molecular results herein, which show clades live sympatrically, it is possible that unidentified morphological apomorphies are correlated with genetic lineages. However, preliminary investigations on our material (Eléaume 2006) show that external morphological characteristics vary among clades. During our study, we were also cognizant of different colour morphs in P. kerguelensis, but their frequencies varied among clades and were not apparently diagnostic of genetic lineages. Thus, the clades most likely represent truly cryptic species.
Using a generalised echinoderm molecular clock (Lessios et al. 1999; McCartney et al. 2000) on the combined average genetic divergences between groups, the deepest split (Clade A + B + C + D vs. E + F) occurred 3.75–4.24 Mya, while the shallowest split (Clade B + C) occurred approximately 537,000–606,000 years ago. This would mean that the putative species contained in Promachocrinus “kerguelensis” diverged well after the onset of the ACC (28–41 Mya, see Lawver and Gahagan 2003; Scher and Martin 2006), but before the last glacial maximum (∼20,000 ya, Huybrechts 2002). If Promachocrinus spp. evolved on the Antarctic continent after the formation of the ACC, and could not subsequently disperse across this barrier, it could explain why, despite comprehensive sampling efforts, Promachocrinus spp. have not been recorded from the Magellanic region (Marr 1963; Speel and Dearborn 1983; but see Mutschke and Rios 2006).
During the last glacial maximum, lowered sea levels, ice sheet grounding and ice flows along the continental shelf were understood to have caused widespread devastation of marine communities (Dell 1972), extending down to shelf limits of 1,000 m (Canals et al. 2000). However, patterns of diversity indicate that it is unlikely that the shelf fauna was eradicated completely at any time during the Cenozoic (Brandt 2005), and the continental slope may have provided refuge during times of glacial maxima. Clarke and Crame (1992) postulated changes in ice-shelf extension drove speciation processes, and perhaps even adaptive radiations. Alternatively, Thatje et al. (2005) suggest that temporal differences in deglaciation may have allowed organisms good at colonizing newly available substrates to move from refuge to refuge around Antarctica, creating the contemporary circumpolar distribution patterns of many taxa. Considering that the pattern of high haplotypic diversity and low nucleotide diversity in Promachocrinus clade A is characteristic of post-bottleneck population expansion (Avise et al. 1984), isolation of Promachocrinus spp. populations during periods of glacial advance is the most likely explanation for this complex of cryptic species.
Currently the number of animal species recorded to date from the Antarctic continental shelf is 3,000–4,000 (Arntz et al. 1997; Clarke and Johnston 2003), but diversity functions predict up to 17,000 (Gutt et al. 2004). These predictive calculations are systematic underestimates due to the spatial scales used, and because they cannot account for morphologically cryptic species (see Gutt et al. 2004). In Antarctic waters, previous studies examining DNA in widespread species have hinted at cryptic species complexes (e.g. Beaumont and Wei 1991; Page and Linse 2002; Held 2003; Held and Wägele 2005; Raupach and Wägele 2006) but sampling schemes have often been limited. Given the complicated climate history of Antarctica, cryptic speciation is likely commonplace.
As with many Antarctic marine species, Promachocrinus “kerguelensis” is currently recognized as having a circum-Antarctic distribution. However, our results raise the question of whether any of the Promachocrinus species-level clades are actually circumpolar. Only one of our clades was found in all four regions (clade A) (Fig. 4), and analyses showed that these populations lacked genetic connectivity. If clade A is distributed around Antarctica, it is not a single, randomly mating population (i.e., panmictic).
Both FST and ϕST analyses revealed significant genetic subdivision for Clade A along the Scotia Arc. This series of islands, seamounts and submerged ridges formed as a consequence of sea floor spreading during the tectonic separation of South America from Antarctica. Although some authors (e.g. Dell 1972; Arntz et al. 2006) hypothesize the Scotia Arc connects populations across the Polar Front, the pairwise FST and ϕST values for clade A indicate a lack of gene flow between the Antarctic Peninsula and South Sandwich Islands. This is surprising given their relative proximity (Fig. 1), and the presence of the ACC and Weddell Sea gyre, which assist in directing water from one region to the other (see also Barnes et al. 2006). Large-scale patterns of dispersal can contrast with present ocean circulation, and might instead reflect past connectivity. However, historical circulation patterns cannot explain lack of gene flow along the Scotia Arc for clade A, as the prevailing ACC has been present longer than the taxon itself. Our results demonstrate restricted gene flow along the Scotia Arc, confirming Picken’s (1985) indirect conclusions that high speciation rates in the Scotia Arc region indicate restricted gene flow.
South Georgia and the South Sandwich Islands are emergent parts of the Scotia Arc, but Bouvetøya Island is a younger (1 Mya, Furnes and Løvlie 1978), exceptionally isolated island to the east, whose marine faunal source populations are unknown. Bouvetøya Island’s general biogeographic affinities appear to lie with South America, although some groups (motile peracarids and non-nudibranch molluscs) show more linkages with the Antarctic Peninsula/Scotia Arc (Arntz et al. 2006). Species-level cheilostome bryozoan communities on Bouvetøya Island showed greatest similarity to the Scotia Arc and Weddell Sea (Barnes 2006). FST and ϕST analyses performed on Promachocrinus clade A showed that Bouvetøya Is. is genetically isolated from the Antarctic Peninsula and from the geographically closer South Georgia and South Sandwich Islands. The haplotypes found on Bouvetøya Is. were all either the assumed ancestral haplotype (A11), or no greater than two mutational steps from it (Fig. 2), which does not assist determining where colonization originated.
In contrast to clade A, clade E consisted of only a single haplotype despite its occurrence over a range of nearly 1,600 km (Fig. 4). Such variation in genetic structure indicates that clades of Promachocrinus have undergone distinct evolutionary trajectories. Possible explanations could include the use of differing refugia during the last glacial maxima. For instance, recolonization from the deep-sea would likely produce different patterns than recolonization from multiple shelf refugia. Alternatively, the maintenance of a single haplotype over a large range (clade E) might indicate high levels of gene flow among populations and/or a rapid colonization and expansion event, or even a selective sweep (Palumbi 1995). A similarly contrasting pattern of gene flow was demonstrated between two Caribbean echinoids, where the lack of a common pattern was attributed to the highly stochastic nature of successful long-distance dispersal by larvae (McCartney et al. 2000), a characteristic dispersal pattern for the Antarctic (Dayton 1989).
The assumed wide range of Promachocrinus “kerguelensis” was influential in forming the prediction that its larvae remained in the water column for long periods (McClintock and Pearse 1987). Although many Antarctic comatulid crinoids are brooders, most Antedonidae are free spawners with short dispersal times (e.g. Florometra serratissima, 5–9 days, Mladenov and Chia 1983; Antedon bifida, 5–8 days, Lahaye 1987; Oxycomanthus japonicus, 1.5–5 days, Holland 1991; Decametra tigrina, 2–12 days, Kohtsuka and Nakano 2005). Given the lack of gene flow between regions for clade A, some Promachocrinus spp. may have a short larval dispersal period, like other comatulid crinoids. McClintock and Pearse (1987) worked in McMurdo Sound, Ross Sea, an area not sampled in this study. This may become important if different clades of Promachocrinus show variability in larval development and subsequent dispersal.
In summary, the widespread Antarctic crinoid Promachocrinus “kerguelensis” is shown to consist of multiple distinct lineages, which likely represent cryptic species. Additional sampling from other areas in Antarctica is needed to assess the full diversity contained within Promachocrinus, and whether any of the clades identified here demonstrate a true circumpolar distribution. Given that P. “kerguelensis” had received greater taxonomic attention than most Antarctic invertebrates and yet still represents multiple cryptic lineages, it is likely that the diversity in Antarctica is vastly underestimated.
References
Allcock AL, Brierley AS, Thorpe JP, Rodhouse PG (1997) Restricted gene flow and evolutionary divergence between geographically separated populations of the Antarctic octopus Pareledone turqueti. Mar Biol 129:97–102
Arntz WE, Rios C (1999) Magellan-Antarctic: ecosystems that drifted apart. Sci Mar 63(Suppl 1):1–518
Arntz WE, Gutt J, Klages M (1997) Antarctic marine biodiversity: an overview. In: Battaglia B, Valencia J, Walton DWH (eds) Antarctic communities: species, structure and survival. Cambridge University Press, Cambridge, pp 3–14
Arntz WE, Thatje S, Linse K, Avila C, Ballesteros M, Barnes DKA, Cope T, Cristobo FJ, de Broyer C, Gutt J, Isla E, López-González P, Montiel A, Munilla T, Ramos Esplá AA, Raupach M, Rauschert M, Rodríguez E, Teixidó N (2006) Missing link in the Southern Ocean: sampling the marine benthic fauna of remote Bouvet Island. Polar Biol 29:83–96
Avise JC, Neigel JE, Arnold J (1984) Demographic influences on mitochondrial DNA lineage survivorship in animal populations. J Mol Evol 20:99–105
Barnes DKA (2006) A most isolated benthos: coastal bryozoans of Bouvet Island. Polar Biol 29:114–119
Barnes DKA, Hodgson DA, Convey P, Allen CS, Clark A (2006) Incursion and excursion of Antarctic biota: past, present and future. Glob Ecol Biogeogr 15:121–142
Beaumont AR, Wei JHC (1991) Morphological and genetic variation in the Antarctic limpet Nacella concinna (Strebel, 1908). J Mollus Stud 57:443–450
Brandt A (2005) Evolution of Antarctic biodiversity in the context of the past: the importance of the Southern Ocean deep sea. Antarct Sci 17:509–521
Canals M, Urgeles R, Calafat AM (2000) Deep sea-floor evidence of past ice streams off the Antarctic Peninsula. Geology 28:31–34
Chen L, DeVries AL, Cheng C-HC (1997) Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish. Proc Natl Acad Sci 94:3811–3816
Clark A (1990) Temperature and evolution. Southern Ocean cooling and the Antarctic marine fauna. In: Kerry KR, Hempel G (eds) Antarctic ecosystems: ecological change and conservation. Springer, Berlin, pp 9–22
Clarke A Crame JA (1992) The Southern Ocean benthic fauna and climate change: a historical perspective. Philos Trans R Soc B 338:299–309
Clarke A, Johnston NM (2003) Antarctic marine benthic diversity. Oceanogr Mar Biol Ann Rev 41:47–114
Clark AH, Clark AM (1967) A monograph of the existing crinoids 1(5). US Nat Mus Bull 82:1–860
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Dayton PK (1989) Interdecadal variation in an Antarctic sponge and its predators from oceanographic climate shifts. Science 245:1484–1486
Dayton PK (1990) Polar benthos. In: Smith WO (ed) Polar oceanography, Part B: chemistry, biology, and geology. Academic Press, San Diego, pp 631–685
Dell RK (1972) Antarctic benthos. Adv Mar Biol 10:1–216
Dell RK (1990) Antarctic Mollusca, with special reference to the Ross Sea. R Soc NZ Bull 27:1–311
Eastman JT, Clarke A (1998) A comparison of adaptive radiations of Antarctic fish with those of non-Antarctic fish. In: di Prisco G, Pisano E, Clark A (eds) Fishes of Antarctica: a biological overview. Springer, Berlin, pp 3–26
Eléaume M (2006) Approche morphométrique de la variabilité phénotypique: conséquences systématiques et évolutives. Application aux crinoïdes actuels (Crinoidea : Echinodermata). Muséum national d’Histoire naturelle, Paris
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech 3:294–299
Furnes H, Løvlie R (1978) An eruptional model for the recent lava flow on Bouvetøya, South Atlantic Ocean. Norsk Polar Skrift 169:103–108
Gray JS (2001) Antarctic marine benthic biodiversity in a world-wide latitudinal context. Polar Biol 24:633–641
Gutt J, Sirenko BI, Smirnov IS, Arntz WE (2004) How many macrozoobenthic species might inhabit the Antarctic shelf? Antarct Sci 16:11–16
Hedgpeth JW (1969) Distribution of selected groups of marine invertebrates in waters south of 35°S latitude. Antarct Map Folio Ser 11:1–7
Held C (2000) Phylogeny and biogeography of serolid isopods (Crustacea, Isopoda, Serolidae) and the use of ribosomal expansion segments in molecular systematics. Mol Phylogenet Evol 15:165–178
Held C (2003) Molecular evidence for cryptic speciation within the widespread Antarctic crustacean Ceratoserolis trilobitoides (Crustacea, Isopoda). In: Huiskes AHL, Giekes WWC, Rozema J, Schorno RML, van der Vies SM, Wolff WJ (eds) Antarctic biology in a global context. Backhuys, Leiden, The Netherlands, pp 135–139
Held C, Wägele J-W (2005) Cryptic speciation in the giant Antarctic isopod Glyptonotus antarcticus (Isopoda: Valvifera: Chaetiliidae). Sci Mar 69:175–181
Helgen LE, Rouse GW (2006) Species delimitation and distribution in Aporometra (Crinoidea: Echinodermata); endemic Australian featherstars. Invertebr Syst 20:395–414
Holland ND (1991) Echinodermata Crinoidea. In: Giese AC, Pearse JS, Pearse VB (eds) Reproduction of marine invertebrates. Boxwood, California, pp 247–299
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Huybrechts P (2002) Sea-level changes at the LGM from ice-dynamic reconstructions of the Greenland and Antarctic ice sheets during the glacial cycles. Q Sci Rev 21:203–231
John DD (1938) Crinoidea. Discov Rep 18:121–222
Knowlton N (1993) Sibling species in the sea. Ann Rev Ecol Syst 24:189–216
Knowlton N (2000) Molecular genetic analyses of species boundaries in the sea. Hydrobiologia 420:73–90
Kohtsuka H, Nakano H (2005) Development and growth of the feather star Decametra tigrina (Crinoidea), with emphasis on the morphological differences between adults and juveniles. J Mar Biol Assoc UK 85:1503–1510
Lahaye MC (1987) Comportement larvaire et ontogenèse postembryonnaire chez la comatule Antedon bifida (Echinodermata, Crinoidea). Thèse. Lab. Biologie Marine, Univ. Libre Bruxelles:1–155
Lawver LA, Gahagan LM (2003) Evolution of Cenozoic seaways in the circum-Antarctic region. Palaeogeo Palaeoclim Palaeoecol 198:11–37
Lessios HA, Kessing BD, Pearse JS (2001) Population structure and speciation in tropical seas: global phylogeography of the sea urchin Diadema. Evolution 55:955–975
Lessios HA, Kessing BD, Robertson DR, Paulay G (1999) Phylogeography of the pantropical sea urchin Eucidaris in relation to land barriers and ocean currents. Evolution 53:806–817
Maddison DR, Maddison WP (2003) MacClade. Sinauer Associates, Inc., Sunderland, Massachusetts
Marr JWS (1963) Unstalked crinoids of the Antarctic Continental Shelf. Philos Trans R Soc B 246:327–379
McCartney MA, Keller G, Lessios HA (2000) Dispersal barriers in tropical oceans and speciation in Atlantic and eastern Pacific sea urchins of the genus Echinometra. Mol Ecol 9:1391–1400
McClintock JB, Pearse JS (1987) Reproductive biology of the common Antarctic crinoid Promachocrinus kerguelensis (Echinodermata: Crinoidea). Mar Biol 96:375–383
Mladenov PV, Chia FS (1983) Development, settling behaviour, metamorphosis and pentacrinoid feeding and growth of the feather star Florometra serratissima. Mar Biol 73:309–323
Mutschke E, Rios C (2006) Spatial distribution and relative abundance of echinoderms from the strait of Magellan, Chile. Cienc Tecnol Mar 29:91–102
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nylander J (2002) MrModeltest. http://www.morphbank.ebc.uu.se/MR.BAYES
Page TJ, Linse K (2002) More evidence of speciation and dispersal across the Antarctic Polar Front through molecular systematics of Southern Ocean Limatula (Bivalvia: Limidae). Polar Biol 25:818–826
Palumbi SR (1992) Marine speciation on a small planet. Trends Ecol Evol 7:114–118
Palumbi SR (1995) Using genetics as an indirect estimator of larval dispersal. In: McEdward L (ed) Ecology of marine invertebrate larvae. CRC, Boca Raton, pp 369–387
Picken GB (1985) Benthic Research in Antarctica: past, present and future. In: Gray JS, Christiansen ME (eds) Marine biology of polar regions and the effects of stress on marine organisms. Wiley, Chichester, pp 167–183
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Rambaut A (2002) Se-Al v2.0a11. University of Oxford, Oxford
Raupach MJ, Wägele J-W (2006) Distinguishing cryptic species in Antarctic Asellota (Crustacea: Isopoda)—a preliminary study of mitochondrial DNA in Acanthaspidia drygalskii. Antarct Sci 18:191–198
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Sará M, Balduzzi A, Barbieri M, Bavestrello G, Burlando B (1992) Biogeographic traits and checklist of Antarctic demosponges. Polar Biol 12:559–585
Scher HD, Martin EE (2006) Timing and climatic consequences of the opening of the Drake Passage. Science 312:428–430
Schneider S, Excoffier L (1999) Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152:1079–1089
Schneider S, Roessli D, Excoffier L (2000) Arlequin: a software for population genetics data analysis. University of Geneva, Geneva
Shaw PW, Arkhipkin AI, Al-Khairulla H (2004) Genetic structuring of Patagonian toothfish populations in the Southwest Atlantic Ocean: the effect of the Antarctic Polar Front and deep-water troughs as barriers to genetic exchange. Mol Ecol 13:3293–3303
Speel JA, Dearborn JH (1983) Comatulid crinoids from the R/V Eltanin cruises in the southern ocean. Antarct Res Ser 38:1–60
Swofford DL (2002) PAUP*. Sinauer, Sunderland, Massachusetts
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Thatje S, Hillenbrand C-D, Larter R (2005) On the origin of Antarctic marine benthic community structure. Trends Ecol Evol 20:534–540
Vaughan DG, Marshall GJ, Connolley WM, Parkinson C, Mulvaney R, Hodgson DA, King JC, Pudsey CJ, Turner J (2003) Recent rapid regional climate warming on the Antarctic Peninsula. Clim Change 60:243–274
Waters JM, O’Loughlin PM, Roy MS (2004) Cladogenesis in a starfish species complex from southern Australia: evidence for vicariant speciation? Mol Phylogenet Evol 32:236–245
Acknowledgments
This study would not have been possible without a huge amount of logistical support from NSF, Raytheon and the crew of the ARSV Laurence M. Gould. Participants in the Halanych/Scheltema cruises and those on ICEFISH 2004 are thanked sincerely for their efforts. Greg Rouse, Lauren Helgen and Chuck Messing were generous in sharing unpublished data, primers, and outgroup information. Financially, this study was supported by NSF grants OPP-9910164 (SGER) and OPP0338087 to R. Scheltema, OPP-0338218 to K. Halanych, and OPP-0132032 to H.W. Deitrich. Sven Thatje and two anonymous reviewers are thanked for their suggestions on improving the manuscript. This study is contribution #23 of the Auburn University (AU) Marine Biology Program, and complies with the current laws of the countries in which the work was performed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J.P. Grassle.
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Wilson, N.G., Hunter, R.L., Lockhart, S.J. et al. Multiple lineages and absence of panmixia in the “circumpolar” crinoid Promachocrinus kerguelensis from the Atlantic sector of Antarctica. Mar Biol 152, 895–904 (2007). https://doi.org/10.1007/s00227-007-0742-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00227-007-0742-9