
Abstract
Marine geographical and ecological barriers often reflect intraspecific genetic discontinuities among populations which may experience different selective pressures and undergo evolutionary divergence. While the phylogeography of marine intertidal invertebrates across the Atlantic/Indian Ocean transition received more attention, the population genetic structures of supralittoral direct developers across such transition area have been poorly investigated. Sandhoppers are supralittoral invertebrates characterised by a direct developmental mode (low dispersal ability), and Talorchestia capensis (Amphipoda, Talitridae) represents the most abundant species of sandhoppers along the South African coasts. To define population structure of T. capensis, we used a mitochondrial marker (the cytochrome oxidase subunit I gene, COX1). T. capensis showed a clean population structure with three main haplogroups genetically well separated, although this separation is not perfectly in line with geographical boundaries described for this area. The presence of separate evolutionary significant units is also confirmed by the shape of mismatch distribution, as well as the p distance values among groups. The overall results confirm the importance of mtDNA to retrieve information on the evolutionary history of species. This study suggests the evidence of a complex-species for this sandhoppers, which have never been considered before, providing fundamental basis for further studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In marine systems, geographical discontinuities are often mirrored by intraspecific genetic discontinuities among populations (Benzie, 1999), occurring where past or present-day barriers to dispersal allow for genetic differentiation and prevent homogenisation due to gene flow within a species (Teske et al., 2011). In the marine realm, oceanographical conditions are the major drivers of phylogeographical breaks (Collin, 2001; Sanford et al., 2003; Tellier et al., 2009; Zakas et al., 2009), although differences in abundance, timing of reproduction, life history traits, population and recruitment dynamics, and physiological traits can also regulate such discontinuities (Broitman et al., 2001; Rivadeneira et al., 2002; Ragionieri et al., 2009; Carson et al., 2010; Kelly & Palumbi, 2010; McQuaid, 2010).
In coastal areas, phylogeographical breaks in marine organisms have mostly been associated with coastal currents and eddies, river discharge and upwelling cells, which all interfere with along-shore dispersal (Pelc et al., 2009; Teske et al., 2011; Brante et al., 2012). In South Africa, coastal species are often divided into genetic lineages distributions of which are linked to the marine biogeographical provinces of southern Africa (the cool temperate, warm temperate, subtropical and tropical provinces, see Teske et al., 2011). The major phylogeographical breaks have been identified at three main localities that, in most cases, coincide with the disjunctions between these four provinces (Fig. 1; Teske et al., 2007; Zardi et al., 2007; Von der Heyden et al., 2008; Teske et al., 2009, 2011). Despite this general pattern, differences in the positions of phylogeographical discontinuities are common among South African marine invertebrates (Teske et al., 2011), especially between planktonic and direct developers (Teske et al., 2007). Such discontinuities may also reflect different Evolutionary Significant Units (ESU’s sensu Moritz, 1994), as well as cryptic species. For example, Teske et al. (2009), investigating the phylogeography of the sand prawn Callianassa krausii along the South African coast, suggested the existence of distinct ESUs, possibly related to environmental discontinuity between the tropical and subtropical regions.

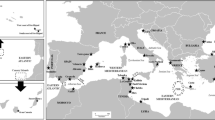
The South African coast, showing model organism, sites of collection, current systems, bioregions and transition zones. PN Port Nolloth, BB Betty’s Bay, GB Gansbaai, SB Still Bay, GOU Gouritzmond, GLE Glentana, PA Port Alfred, PSJ Port St. Johns. Black-dashed circles represent the phylogeographical transition zones (Teske et al., 2011): Agulhas, at the transition between the Atlantic and Indian Oceans, Cape Bioregion; Algoa Bay to Wild Coast in the Agulhas Bioregion and St Lucia, in the Subtropical Bioregion. Bioregions are reported in the grey scale bar at the bottom of the figure. CT cool-temperate bioregion, C Cape Bioregion, WT warm-temperate bioregion, ST subtropical bioregion
Sandhoppers, supralittoral amphipods of the family Talitridae, are direct developers (i.e. no free-swimming larval form, eggs are brooded and hatch as juveniles with an adult-like form), showing low active dispersal ability (Dahl, 1946; Wildish, 1970). Due to the absence of a dispersive stage, through which active dispersal occurs or by which passive dispersal is facilitated by oceanographical conditions, direct developers generally show low levels of population connectivity, reduced mixing among local populations and, consequently, clear population genetic structure (Pavesi et al., 2013; Fanini & Lowry, 2014). Notwithstanding, direct-developing species can potentially rely on rafting as the main long distance dispersal mechanism (Thiel & Gutow, 2005a, b). However, such a mechanism has rarely been demonstrated for talitrids (Persson, 2001); in fact, Platorchestia platensis (Krøyer, 1845) is the only talitrid with strong evidence of rafting as a dispersal mechanism (Simpson, 2011).
Talorchestia capensis (Dana, 1853) is a talitrid species showing a widespread distribution along the South African coast (Baldanzi et al., 2013), encompassing four South African bioregions (Lombard, 2004) characterised by differing environments. Furthermore, individuals of T. capensis from locations found within this range showed different thermal tolerances, likely shaped by local environmental conditions (Baldanzi et al., 2015). Both these biogeographical and ecophysiological characteristics of T. capensis suggest potential genetic differentiation among separated populations. Given that, we investigated the population genetic structure and demographical history of T. capensis using mitochondrial DNA as a molecular marker. We expected that T. capensis should show clear population structure, as reported for many other sandhoppers (Pavesi et al., 2011, 2012, 2013). We envisage that Talorchestia capensis should show an intraspecific phylogeny associated with defined geographical areas, but, nonetheless, such genetic breaks would not coincide with South Africa’s bioregions, in agreement with Teske et al. (2011) assertion for local species with low dispersal ability.
Materials and methods
Study area and animal collection
Talorchestia capensis specimens were collected during 2012–2013 from 8 locations along the South African coast, encompassing four bioregions (Fig. 1; Table 1). Localities within the Cape region (from Cape Point to Agulhas: BB and GB populations, Table 1) are considered to be in a transition area between the warm and cool temperate regions (Teske et al., 2011). PSJ is the only location within the subtropical region and it represents the eastern limit of the spatial distribution of T. capensis. Locations were chosen on the basis of accessibility and the presence of animals. Figure 1 shows the locations, bioregions and the South African current systems, the Agulhas and Benguela Currents, which dominate the south-east and the south-west coasts, respectively. Between 6 and 15 individuals per location were collected by hand and stored in 100% ethanol (Table 1).
DNA extraction, amplification and sequencing
DNA was extracted from the telson of the animals following a CTAB (cetyltrimethyl ammonium bromide) method (Doyle & Doyle, 1987). A portion of the mitochondrial cytochrome c oxidase subunit I gene (mtDNA COX1) was amplified using polymerase chain reaction (PCR). The forward primer CrustCOIF 5′ and reverse primer PeracCOIR 5′ (Teske et al., 2007) were used to amplify all the specimens for all populations. The PCR profile comprised an initial denaturation step of 3 min at 94°C, followed by 35 cycles of denaturation (30 s at 94°C), annealing (45 s at 48–50°C) and extension (75 s at 72°C); and a final extension step of 10 min at 72°C. PCR products were purified and cleaned with a DNA clean up kit (MSB® Spin PCRapace, Stratec®), cycle sequenced both in the forward and reverse direction using the BigDye Terminator v.3.1 Cycle Sequencing kit (Applied Biosystems) and sequenced on an ABI 3100 genetic analyser. The sequences were edited using the software SEQUENCHER v4.8 (Gene Code Corporation, Ann Arbor, MI, USA), and edited sequences were aligned using the MUSCLE algorithm in MEGA v5.05 (Tamura et al., 2011).
Data analysis
The model of DNA substitution that best fitted our data was chosen using the software MODELTEST 3.6 (Posada & Crandall, 1998). Two methods of phylogenetic inference were applied for reconstructing phylogenetic relationships among haplotypes: Maximum Likelihood (ML), using the software PAUP*4b10 (Swofford, 2002) and Bayesian analysis (BI) as implemented in MrBayes v.3.2.6 (Huelsenbeck & Ronquist, 2001). For ML, the parameters of the suggested model of evolution were implemented in PAUP. The ML starting tree was obtained via stepwise addition and replicated 10 times, with each replicate starting with a random input order of sequences. Clade support was obtained from 1000 bootstrap replicates (Felsenstein, 1985). The BI trees were calculated using the suggested model of evolution. The Bayesian analysis was run with four MCMC (Markov chain Monte Carlo) for 2,000,000 generations, saving a tree every 500 generations (with a corresponding output of 4000 trees). The −lnL converged on a stable value between 5,000 and 10,000 generations (“burnin phase”). The first 12,000 generations were thus not included in the analysis to avoid the possibility of including random and suboptimal trees. The posterior probabilities of the phylogeny were determined by constructing a 50% majority-rule consensus of the remaining trees. Consensus trees were obtained using the “sumpt” option in MrBayes. Sequences of the talitrids Orchestia uhleri and Platorchestia monodi were added as outgroups (GenBank accession nos. AY152751 and AB741727, respectively).
An additional Median-Joining (MJ) haplotype network (Bandelt et al., 1999) of mtDNA haplotypes was drawn to identify groupings of individuals among the populations and to examine haplotype frequencies and distributions (networks were constructed for the full dataset). This implemented a Maximum Parsimony calculation to eliminate non-parsimonious links among haplotypes (Polzin & Daneschmand, 2003). The geographical distribution of the individuals/haplotypes was considered against the resulting network. The network was generated using the software NETWORK v4.6 (Fluxus Technology, Ltd; www.fluxus-engineering.com).
The existence of population structure was assessed by one-way AMOVA (Excoffier et al., 1992), as implemented in Arlequin v3.5 (Excoffier et al., 2005). ΦST values (Wright, 1951) were calculated based on genetic distances, assessed by the Tajima & Nei (1984) method, which is recommended for unequal nucleotide frequencies as recorded in our dataset (Tajima & Nei, 1984). Significance levels of pairwise ΦST under the null hypothesis of no differentiation among populations were tested using a non-parametric permutation approach (10,000 permutations of haplotypes among populations, Excoffier et al., 1992). Two additional two-way AMOVAs, with populations grouped as three groups, were performed according to two alternative biogeographical hypotheses. Hypothesis 1 took into account previous phylogeographical studies carried out on southern African coastal invertebrates (see review by Teske et al., 2011 and references therein), which revealed two main phylogeographical breaks at Cape Agulhas and Algoa Bay (Fig. 1). Thus, populations were grouped as follows (see Table 1 for locality/population abbreviations): a south-western group (S-W), including populations PN, GB and BB; a southern group (S), with SB, GOU and GLE populations; and a south-eastern group (S-E), including PA and PSJ populations. Alternatively, Hypothesis 2 was based on the results we obtained from the phylogenetic inferences and differed from Hypothesis 1 with populations GOU and SB included in group S-W instead of group S.
Additionally, partitioning of genetic variation in relation to geography was tested by applying a Spatial Analysis of Molecular Variance (SAMOVA), as implemented in the software SAMOVA 2.0 (Dupanloup et al., 2002), with 1,000 permutations and setting the number of initial conditions to 100 as recommended by Dupanloup et al. (2002). We used the new SAMOVA option of assigning populations to K groups based on sequence similarity, without constraint for the geographical composition of groups. The most likely structure corresponds to the partition of populations maximizing among-group variation as measured by the AMOVA ΦCT statistic (Excoffier et al., 1992). The number of groups (K) of populations ranged from 2 to 7. The K with the highest ΦCT represents the best number of groups and the best population configuration. On the other hand, higher K configurations with single population groups cannot provide the group structure (Heuertz et al., 2004). Therefore, in case of similar ΦCT associated with different K values, the lowest K was chosen as the optimal clustering solution.
The demographical history of the metapopulation, as well as of each population and the identified lineages (see below), was reconstructed by applying mismatch distribution analyses (i.e. the distribution of the observed number of differences between pairs of haplotypes) and two neutrality tests, Tajima’s (1989) D test and Fu’s (Fu & Li, 1993; Fu, 1997) Fs test, as implemented in ARLEQUIN 3.5. Populations at demographical equilibrium are expected to show a multimodal distribution of haplotype frequencies, while populations having recently passed through a demographical expansion or a range expansion are predicted to have a unimodal distribution (Rogers & Harpending, 1992). ARLEQUIN 3.5 compares the observed distribution with that expected under a model of population expansion [based on the Harpending’s raggedness index (rg), Rogers, 1995], using a parametric bootstrap approach (10,000 replicates) under the null hypothesis of population expansion.
We calculated the mean genetic p-distance (±SE) among inferred groups with MEGA 3.1 (Kumar et al., 2004), with a simple nucleotide model (number of substitutions), a homogeneous pattern of mutation among lineages and uniform rate among sites. Then, the mean divergence time between groups was estimated by applying the crustacean mutation rate per site per year, defined by Knowlton et al. (1993) (i.e. approximately 2.5%) and already used in other studies of talitrids (see Pavesi et al., 2012, 2013), with the standard error of divergence time being the standard error of the mean genetic p-distance divided by the rate.
Results
The COX1 fragment had an A-T rich nucleotide composition (C = 17.5%; A = 26%; T = 39.3%; G = 17.2%), as reported for other arthropod mitochondrial DNA (Simon et al., 1994). A total of 101 variable sites were obtained from 80 individuals sequenced for a 585 bp fragment of COX1, resulting in 48 haplotypes. The overall average number of nucleotide/base pair differences between haplotypes was 3.76 ± 1.85 (Table 1). Most of the observed mutations were in the 3rd codon positions and were synonymous with respect to amino acid translations. Only 19 mutations induced amino acid substitutions. Inclusion of the two outgroup haplotypes increased the variable sites to 169, of which 104 were parsimony informative and 24 induced amino acid substitutions.
The selected model of DNA substitution was the HKY85 model (Hasegawa et al., 1985), with unequal substitution rates and a gamma distribution shape parameter (α of 0.221). This model was used in the ML and BI analysis. Both phylogenetic inference methods resulted in highly congruent topologies and the slight differences did not affect the general definition of the lineages (Fig. 2). The phylogenetic inference showed three main lineages of T. capensis: a South-West (S-W) lineage, including individuals from the west coast (PN), the Cape region (GB and BB) and the south coast (SB and GOU); a South (S) lineage, including individuals from the south coast (GLE) and individuals from GOU and SB (other individuals from these localities clustered within S-W); and a South-East (S-E) lineage (PA and PSJ), which also included individuals from the S–W (SB and GOU) and S lineages (GLE) (Fig. 2). Both ML and BI supported most main clades well, with the ML calculation generally giving the highest support (Fig. 2). Overlap among the three lineages was from SB to GLE on the south coast, as individuals with haplotypes from each of the three respective lineages were found in this area (Fig. 2).

Maximum Likelihood phylogenetic tree of mtDNA COX1 haplotypes of T. capensis. Branch lengths are in the same units as the evolutionary distances. The percentages of bootstrap replicates in which the associated taxa clustered together are shown on the nodes (minimum accepted threshold: 70%). Maximum Likelihood bootstrap percentages and Bayesian inference posterior probabilities are shown as follows: ML/BI. The left upper quadrant shows the South African coastline with the distribution of the clades. Solid lines are the effective locations of the clade; dashed lines represent the expected locations but from which sequences were not retrieved. The terminals represent individuals. Refer to Fig. 1 for location codes
The MJ haplotype network (Fig. 3) also showed three divergent lineages, confirming the phylogenetic inference (Fig. 2). In the network, the S-W lineage showed the highest number of shared haplotypes (29 individuals sharing five different haplotypes), while the S-E lineage showed nine individuals sharing three haplotypes. In the S lineage, only one haplotype was shared among three individuals.). Moreover, individuals from GOU, GLE and SB populations clustered within different lineages.

MJ haplotype network of mtDNA COX1 region of T. capensis. Haplotypes are shown as pie charts in which size is proportional to the number of individual sharing that haplotype. Numbers of individuals greater than one are reported next to each shared haplotype. Haplotype colours are shown in the legend on the left of the graph, each colour corresponding to different populations and geographical origins. Numbers of mutational steps are reported for the three main branches forming the three lineages. All branches’ lengths are proportional to the number of mutational steps
The one-way AMOVA provided evidence of strong population structure among all populations (ΦST = 0.66, P < 0.001). All the pairwise ΦST values (data not shown) were significant (P < 0.05), except the following comparisons: GB versus BB, GOU versus SB, and PSJ versus PA. The results of the two additional AMOVAs (Table 2) suggested that Hypothesis 2 is a valid alternative to Hypothesis 1 as it better explains the partitioning of genetic variation among groups, as it recorded the higher ΦCT value (the among-group component of variance). Nonetheless, both the hypotheses seem appropriate to describe the haplotype variation reported.
The SAMOVA analysis resulted in statistically significant ΦCT values at K = 2–7. The best option of population grouping, reporting a ΦCT value of 0.70, corresponded to K = 3 and reflected the geographical distribution of the AMOVA’s Hypothesis 2 (Table 2). The same ΦCT value was also associated with K = 6, with a further separation of lineage S-W into four distinct groups (PV vs BB + GB vs GOU vs SB); since this K option increased the number of single population groups to 4, it could not provide the appropriate group structure as discussed in Heuertz et al. (2004). Indeed, 70.2% of the total variation at the among group level was explained with K = 3 and K = 6, while 61.6, 69.6, 69.9, and 68.4% was explained with K = 2, K = 4, K = 5 and K = 7, respectively.
The mismatch distribution calculated for the overall metapopulation (Fig. 4A) shows three groups of pairwise nucleotide differences, in line with what reported from the network and the phylogenetic tree. Group III, which is completely separated, consists of the classes of 32–45 nucleotide differences, while the separation between Group I and II is less evident and occurs approximately around 20 nucleotide differences. Reconstructing the mismatch distribution for the three lineages, we did not observe the same pattern of the metapopulation: in fact, the S-W and S lineages have a bimodal distribution with a clear disjunction between the two peaks (Fig. 4B, C), a likely indication of the occurrence of two distinct lineages in each of these two geographical areas. Conversely, the S-E lineage had a multimodal mismatch distribution (Fig. 4D), as expected for populations at equilibrium.

Mismatch distribution. Observed mismatch distribution for A the total metapopulation, B group S-W, C group S and D group S-E. In diagram A, major groups of pairwise nucleotide differences are numbered
The mismatch distribution analyses reported negative and significant Fu’s F S value for the S-W lineage, while significant Tajima’s D value for the S lineage (Table 1). Moreover, no raggedness index was significant, i.e. it was not possible to reject the null hypothesis of a deviation from equilibrium for any of the three lineages and the overall metapopulation (Table 1).
The mean genetic p-distances shows that the three haplogroups are genetically well separated from each other (S-W vs S-E: 6.2 ± 0.8; S-W vs S: 6.4 ± 0.9; S vs S-E: 4.3 ± 0.7). We, therefore, applied the molecular clock using the crustacean CoxI mutation rate per site per year defined by Knowlton et al. (1993) (i.e. approximately 2.5%), obtaining the following divergence times: between S-W and S-E 2.48 ± 0.32 MYA; between S-W and S 2.56 ± 0.36 MYA; and between S-E and S 1.72 ± 0.28 MYA.
Discussion
The present study used mtDNA to investigate the genetic structure and demographical history of the sandhopper Talorchestia capensis along the coast of South Africa. Overall, our genetic diversity and historical demographical results show high levels of genetic divergence among populations of T. capensis and a clear geographical distribution of haplotypes. These results are in line with other studies on talitrid species, especially in the Mediterranean region (Pavesi et al., 2011, 2012, 2013; Fanini & Lowry, 2014). In particular, T capensis is characterised by a strong genetic structure with the presence of a minimum of three genetically well-separated lineages (with the highest variation explained among groups). Such a result agrees with the general expectation that, sandhoppers, as direct developers (which lack an active planktonic stage), show low dispersal ability, resulting in low levels of genetic exchange (Kelly & Palumbi, 2010).
The strong statistical support for among-group variation (in the AMOVA and SAMOVA tests), the shape of the network and the phylogenetic inference, as well as the high p-distance values between the separated groups suggest the existence of at least three well diverged lineages. Also, the shape of the mismatch distribution recorded for the pooled sample suggests the presence of distinct haplogroups, reciprocally separated. This genetic separation among groups that seems to have occurred far in the past (approximately between 2.5 and 1.5 MYA) suggests the presence of at least three Evolutionarily Significant Units (ESUs, sensu Moritz, 1994). This separation may therefore reflect allopatric divergence, as reported for COX1 profiles in a wide range of taxa (Hebert et al., 2003). Ragionieri et al. (2009), investigating the biogeography of the mangrove crab Neosarmatium meinerti, found the presence of a species-complex, providing genetic and morphological evidence for the existence of four pseudo-cryptic species. Talorchestia capensis showed three different groups with a higher number of mutations (approximately 25–35) in a comparable fragment length (4–5 mutations in a 600 bp long fragment) to those of Ragionieri et al. (2009). Thus, the three lineages of T. capensis may be considered as forming a species-complex, even if we recognise the use of a single locus as a limitation for assigning them a higher taxonomic level (see below). Furthermore, a recent morphological investigation of T. capensis from the same populations did not reveal the occurrence of discriminant morphological traits in specimens belonging to the three haplogroups (Lowry & Baldanzi, submitted), adding a certain degree of uncertainty when assessing taxonomic levels for this sandhopper.
The distributions of the three ESUs/putative species of T. capensis are generally geographically well defined (although some uncertainty remains due to zones of overlap): one is present on the south coast and colonised those habitats first; two units on the south-west and south-east coasts, respectively, are both more recently generated. The three putative species overlap in an area located between Still Bay, Gouritsmond and Glentana (Western Cape, South Africa), suggesting that both the South-West and the South-East lineages maybe expanding their ranges towards the south. An overlap among different ESUs is also suggested from the mismatch distributions recorded for the S-W and S groups, which show two modal peaks separated from each other by not less than 20 mutations, as suggested by Alvarado Bremer et al. (2005). Potential overlap could occur on the margins between the South and the South-East units, but this is difficult to address, given the low number of individuals available (especially in the gap between GLE and PA).
The geographical pattern found in the present study (i.e. SAMOVA) is consistent with most of the work carried out on marine invertebrates of southern Africa (reviewed by Teske et al., 2011), which found that the phylogeography of coastal species is largely linked to the marine biogeographical provinces of southern Africa (Teske et al., 2009). The current study, however, found a break separating the South and the South-West populations from the South-East population of T. capensis along a stretch of coast approximately between Witsand and Gouritsmond (Western Cape, South Africa). This represents a slightly different result compared to the work mentioned above, which proposed the Agulhas region as a common geographical break for most of the invertebrates examined (Teske et al., 2011). Complex mechanisms, such as life history traits, timing of reproduction, changes in abundance and adaptation to local environmental conditions, can be as important as physical barriers in determining these coastal phylogeographical breaks (Carson et al., 2010; Brante et al., 2012; Teske et al., 2013). The abundance of T. capensis, as for sandhoppers in general (Pavesi et al., 2013), is highly dependent on the morphodynamic state of their habitats (i.e. beach slope; Baldanzi et al., 2013). Thus, optimal species-specific environmental conditions could explain the unusual break found in this study. T. capensis shows a decrease in abundance from the Cape region to the south-east coast, likely due to concomitant decrease of available resources (Baldanzi et al., 2013). Such a decrease in abundance, however, could be attributed to the phylogeographical breaks found between Witsand and Gouritsmond (in this study), as proposed by Broitman et al. (2001) and Rivadeneira et al. (2002) for the Chilean coast.
A more taxon-specific explanation for the break found in this study could be the different dispersal potentials of sandhoppers living in ephemeral habitats and those who live in open and exposed sandy shores (Pavesi et al., 2012). For instance, if sandhoppers’ dispersal is facilitated by rafting (Persson, 2001; Fanini & Lowry, 2014), habitats close to estuaries or with a greater amount of wrack and macroalgae (e.g. the Cape sites, characterised by large amounts of kelp wrack, and those sites close to estuaries like Still Bay, Witsand and Gouritsmond) will host highly connected populations. From Gouritsmond eastwards (along the south-east coast of South Africa), the presence of many exposed sandy shores with low amounts of macroalgae (Anderson et al., 2007; SB pers. obs.) may suggest Gouritsmond as a natural/ecological boundary, separating the South–West from the South and South–East populations. Sampling at a higher resolution along the south coast and analysing nuclear genetic markers would certainly clarify the genetic structure of these populations.
The low number of sampling sites between the South and the South-East populations create difficulty in identifying the location of the phylogeographical break between the two lineages. This region, however, may have phylogeographical break anywhere along its extension (Teske et al., 2015). The high number of mutations between the two lineages, however, shows a clear discontinuity, which could be located in the transition area between Algoa Bay and the Wild Coast, as suggested by several authors (reviewed by Teske et al., 2011, 2015; Mmonwa et al., 2015). Greater resolution in the selection of sampling sites is, however, recommended for future work dealing with phylogeographical discontinuities between the two lineages.
Our results are based on a single maternal-inherited marker and are not supported by morphological investigations (Lowry & Baldanzi, submitted). Given this, we are aware that before taxonomical conclusions can be reached, this study would need to be complemented with other studies based on nuclear and/or ecological data for depicting recent and historical processes as well as potential adaptive differences between populations (Fraser & Bernatchez, 2001; Teske et al., 2015). Even according to Moritz (1994), ESUs are designated on the basis of reciprocal monophyly at mitochondrial markers and significant divergence in nuclear loci. Nevertheless, the level of sequence divergence obtained among our phylogeographical groups typically would require millions of years of separation (Fraser & Bernatchez, 2001). Thus, we believe these evolutionary lineages are presumably to be confirmed with nuclear data, since discordance between mitochondrial and nuclear loci are generally expected for recently diverged taxa (Shaw, 2002). Moreover, for T. capensis we can reasonably exclude sex-biased dispersal asymmetry (as reported for species with known female/male philopatry), another reason causing conflicts between mitochondrial and nuclear markers (Toews & Brelsford, 2012).
In conclusion, this work contributes to the understanding of the phylogeography and demographical history of separated populations of T. capensis, a highly abundant South African coastal invertebrate (Baldanzi et al., 2013). The phylogeographical barriers which contributed to the highly defined population structure of T. capensis may be attributed to differences in the local environment, rather than real geographical barriers. It is, however, recommended that further researches consider the possibility that the clear structure of the populations reported in this study might represent cryptic speciation.
References
Alvarado Bremer, J. R., J. Viñas, J. Mejuto, B. Ely & C. Pla, 2005. Comparative phylogeography of Atlantic bluefin tuna and swordfish: the combined effects of vicariance, secondary contact, introgression, and population expansion on the regional phylogenies of two highly migratory pelagic fishes. Molecular Phylogenetic and Evolution 36: 169–187.
Anderson, R. J., A. Rand, M. D. Rothman, A. Share & J. J. Bolton, 2007. Mapping and quantifying the South African kelp resource. Scientific Journal of Marine Science 29: 369–378.
Baldanzi, S., C. D. McQuaid, S. Cannicci & F. Porri, 2013. Environmental domains and range-limiting mechanisms: testing the abundant centre hypothesis using Southern African sandhoppers. PLoS ONE 8: e54598.
Baldanzi, S., N. Weidberg, M. Fusi, S. Cannicci, C. D. McQuaid & F. Porri, 2015. Contrasting environments shape thermal physiology across the spatial range of the sandhopper Talorchestia capensis. Oecologia 179: 1067–1078.
Bandelt, H.-J., P. Forster & A. Röhl, 1999. Median joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution 16: 37–48.
Benzie, J. A. H., 1999. Genetic structure of coral reef organisms: ghosts of dispersal past. American Zoologist 39: 131–145.
Brante, A., M. Fernández & F. Viard, 2012. Phylogeography and biogeography concordance in the marine gastropod Crepipatella dilatata (Calyptraeidae) along the southeastern Pacific coast. Journal of Heredity 103: 630–637.
Broitman, B. R., S. A. Navarrete, F. Smith & S. D. Gaines, 2001. Geographic variation of southeastern Pacific intertidal communities. Marine Ecology Progress Series 224: 21–34.
Carson, H. S., M. P. López-Duarte, L. Rasmussen, D. Wang & L. A. Levin, 2010. Reproductive timing alters population connectivity in marine metapopulations. Current Biology 20: 1926–1931.
Collin, R., 2001. The effects of mode of development on phylogeography and population structure of North Atlantic Crepidula (Gastropoda: Calyptraeidae). Molecular Ecology 10: 2249–2262.
Dahl, E., 1946. The Amphipoda of the Sound. Part 1. Terrestrial Amphipoda. Acta University Lund, N.F. Adv. 242, 1–53.
Doyle, J. J. & J. L. Doyle, 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11–15.
Dupanloup, I., S. Schneider & L. Excoffier, 2002. A simulated annealing approach to define the genetic structure of populations. Molecular Ecology 11: 2571–2581.
Excoffier, L., P. E. Smouse & J. M. Quattro, 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Excoffier, L., G. Laval & S. Schneider, 2005. ARLEQUIN ver. 3.0: an integrated software package for population genetics data analysis. Evolutionary Bioinformatic Online. 1: 47–50.
Fanini, L. & J. Lowry, 2014. Coastal talitrids and connectivity between beaches: a behavioural test. Journal of Experimental Marine Ecology and Biology 457: 120–127.
Felsenstein, J., 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791.
Fraser, D. J. & L. Bernatchez, 2001. Adaptive evolutionary conservation: towards a unified concept for defining conservation units. Molecular Ecology 10: 2741–2752.
Fu, Y.-X., 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925.
Fu, Y.-X. & W.-H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709.
Hasegawa, M., H. Kishino & T. Yano, 1985. Dating the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22: 160–174.
Hebert, P. D. N., A. Cywinska, S. L. Ball & J. R. deWaard, 2003. Biological identifications through DNA barcodes. Proceedings of the Royal Society, London B 270: 313–321.
Heuertz, M., S. Fineschi, M. Anzidei, R. Pastorelli, D. Salvini, L. Paule, et al., 2004. Chloroplast DNA variation and postglacial recolonization of common ash (Fraxinus excelsior L.) in Europe. Molecular Ecolology 13: 3437–3452.
Huelsenbeck, J. P. & F. Ronquist, 2001. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755.
Kelly, R. P. & S. R. Palumbi, 2010. Genetic structure among 50 species of the northeastern Pacific Rocky Intertidal Community. PLoS ONE 5: e8594.
Knowlton, N., L. A. Weight, L. A. Solorzano, D. K. Mills & E. Berming-ham, 1993. Divergence in proteins, mitochondrial DNA, and reproductive compatibility across the isthmus of Panama. Science 260: 1629–1632.
Kumar, S., K. Tamura & M. Nei, 2004. MEGA3: integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Briefing in Bioinformatics 5: 150–163.
Lombard, A. T., 2004. Marine Component of the National Spatial Biodiversity Assessment for the Development of South Africa’s National Biodiversity Strategic and Action Plan. National Botanical Institute: 101 pp.
McQuaid, C. D., 2010. Marine connectivity: timing is everything. Current Biology 20: 938–940.
Mmonwa, K. L., P. R. Teske, C. D. McQuaid & N. P. Barker, 2015. Historical demography of southern African patellid limpets: congruence of population expansions, but not phylogeography. African Journal of Marine Science 37: 11–21.
Moritz, C., 1994. Defining “Evolutionarily Significant Units” for conservation. Trends in Ecology & Evolution 9: 373–375.
Pavesi, L., E. De Matthaeis, R. Tiedemann & V. Ketmaier, 2011. Temporal population genetics and COI phylogeography of the sandhopper Macarorchestia remyi (Amphipoda: Talitridae). Zoological Studies 50: 220–229.
Pavesi, L., A. Deidun, E. De Matthaeis, R. Tiedemann & V. Ketmaier, 2012. Mitochondrial DNA and microsatellites reveal significant divergence in the beachflea Orchestia montagui (Talitridae: Amphipoda). Aquatic Science 74: 587–596.
Pavesi, L., R. Tiedemann, E. De Matthaeis & V. Ketmaier, 2013. Genetic connectivity between land and sea: the case of the beachflea Orchestia montagui (Crustacea, Amphipoda, Talitridae) in the Mediterranean Sea. Zoological Frontiers 10: 21.
Pelc, R. A., R. R. Warner & S. D. Gaines, 2009. Geographical patterns of genetic structure in marine species with contrasting life histories. Journal of Biogeography 36: 1881–1890.
Persson, L. E., 2001. Dispersal of Platorchestia platensis (Kröyer) (Amphipoda: Talitridae) along Swedish coasts: a slow but successful process. Estuaries Coastal and Shelf Science 52: 201–210.
Polzin, T. & S. V. Daneschmand, 2003. On Steiner trees and minimum spanning trees in hypergraphs. Operation Research Letters 31: 12–20.
Posada, D. & K. A. Crandall, 1998. Modeltest: testing the model of DNA substitution. Bioinformatics 14: 817–818.
Ragionieri, L., S. Fratini, M. Vannini & D. Christoph, 2009. Phylogenetic and morphometric differentiation reveal geographic radiation and pseudocryptic speciation in a mangrove crab from the Indo-West Pacific. Molecular Phylogenetics and Evolution 52: 825–834.
Rivadeneira, M. M., M. Fernàndez & S. A. Navarrete, 2002. Latitudinal trends of species diversity in rocky intertidal herbivore assemblages: spatial scale and the relationship between local and regional species richness. Marine Ecology Progress Series 245: 123–131.
Rogers, A., 1995. Genetic evidence for a Pleistocene population explosion. Evolution 49: 08–615.
Rogers, A. R. & H. Harpending, 1992. Population growth makes waves in the distribution of pairwise genetic distances. Molecular Biology and Evolution 9: 552–569.
Sanford, E., M. S. Roth, G. C. Johns, J. P. Wares & G. N. Somero, 2003. Local selection and latitudinal variation in a marine predator-prey interaction. Science 300: 1135–1137.
Shaw, K. L., 2002. Conflict between nuclear and mitochondrial DNA phylogenies of a recent species radiation: what mtDNA reveals and conceals about modes of speciation in Hawaiian crickets. Proceedings of the National Academy of Sciences of the United States of America 99: 16122–16127.
Simon, C., F. Frati, A. Beckenbach, B. Crespi, H. Liu & P. Flook, 1994. Evolution, weighting and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Annals of the Entomological Society of America 87: 651–701.
Simpson, R., 2011. The invasive biology of the talitrid amphipod Platorchestia platensis in North West Europe. The Plymouth Student Scientist 4: 278–292.
Swofford, D. L., 2002. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Sinauer Associates, Sunderland, MA.
Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Tajima, F. & M. Nei, 1984. Estimation of evolutionary distance between nucleotide sequences. Molecular Biology and Evolution 1: 269–285.
Tamura, K., D. Peterson, N. Peterson, G. Stecher, M. Nei & S. Kumar, 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Biology and Evolution. doi:10.1093/molbev/msr121.
Tellier, F., A. P. Meynard, J. A. Correa, S. Faugeron & M. Valero, 2009. Phylogeographic analyses of the 30- S south-east Pacific biogeographic transition zone establish the occurrence of a sharp genetic discontinuity in the kelp Lessonia nigrescens: vicariance or parapatry? Molecular Phylogenetics and Evolution 53: 679–693.
Teske, P. R., I. Papadopoulos, G. I. Zardi, C. D. McQuaid, M. T. Edkins, C. L. Griffiths & N. P. Barker, 2007. Implications of life history for genetic structure and migration rates of southern African coastal invertebrates: planktonic, abbreviated and direct development. Marine Biology 152: 697–711.
Teske, P. R., H. Winker, C. D. McQuaid & N. P. Barker, 2009. A tropical/subtropical biogeographic disjunction in southeastern Africa separates two evolutionary significant units of an estuarine prawn. Marine Biology 156: 1265–1275.
Teske, P. R., S. von der Heyden, C. D. McQuaid & N. P. Barker, 2011. A review of marine phylogeography in southern Africa. South African Journal of Science 107: 43–53.
Teske, P. R., G. I. Zardi, C. D. McQuaid & K. R. Nicastro, 2013. Two sides of the same coin: extinctions and originations across the Atlantic/Indian Ocean boundary as consequences of the same climate oscillation. Frontiers in Biogeography 5: 48–59.
Teske, P. R., S. Bader & T. R. Golla, 2015. Dispersal against an ocean current. Marine Ecology Progress Series 539: 153–163.
Thiel, M. & L. Gutow, 2005a. The ecology of rafting in the marine environment. I. The floating substrata. Oceanography and Marine Biology Annual Review. 42: 181–263.
Thiel, M. & L. Gutow, 2005b. The ecology of rafting in the marine environment. II. The rafting organisms and community. Oceanography and Marine Biology Annual Review. 43: 279–418.
Toews, D. P. L. & A. Brelsford, 2012. The biogeography of mitochondrial and nuclear discordance in animals. Molecular Ecology 21: 3907–3930.
Von der Heyden, S., K. Prochazka & R. C. K. Bowie, 2008. Significant population structure and asymmetric gene flow patterns amidst expanding populations of Clinus cottoides (Perciformes, Clinidae): application of molecular data to marine conservation planning in South Africa. Molecular Ecology 17: 4812–4826.
Wildish, D. J., 1970. Some factors affecting the distribution of Orchestia Leach in estuaries. Journal of Experimental Marine Biology and Ecology 5: 276–284.
Wright, S., 1951. The genetical structure of populations. Annual of Eugenetics 15: 323–354.
Zakas, C., J. Binford, S. A. Navarrete & J. P. Wares, 2009. Restricted gene flow in Chilean barnacles reflects an oceanographic and biogeographic transition zone. Marine Ecology Progress Series 394: 165–177.
Zardi, G. I., C. D. McQuaid, P. R. Teske & N. P. Barker, 2007. Unexpected genetic structure of indigenous (Perna perna) and invasive (Mytilus galloprovincialis) mussel populations in South Africa. Marine Ecology Progress Series 337: 135–144.
Acknowledgments
The authors are extremely grateful to Prof Christopher D. McQuaid for his precious comments on the first draft of this paper and for funding through the South African Research Chairs Initiative of the Department of Science and Technology. The authors extend their gratitude to Dr. Peter Teske for his advice on data analysis, Dr. Stefano Cannicci and Dr. Francesca Porri for their contribution on an early draft of this paper. SB is particularly grateful to Dr. C. Kelly for his help in the laboratory. The authors finally thank two anonymous reviewers who contributed to improve the manuscript. The work is based upon research supported by the South African Research Chairs Initiative of the Department of Science and Technology and the National Research Foundation of South Africa (NRF).
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Diego Fontaneto
Rights and permissions
About this article

Cite this article
Baldanzi, S., Gouws, G., Barker, N.P. et al. Molecular evidence of distinct evolutionary units in the sandhopper Talorchestia capensis (Amphipoda, Talitridae) along South African coasts. Hydrobiologia 779, 35–46 (2016). https://doi.org/10.1007/s10750-016-2797-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-016-2797-4