
Abstract
Bone mineral carbonate content assessed by vibrational spectroscopy relates to fracture incidence, and mineral maturity/ crystallinity (MMC) relates to tissue age. As FT-IR and Raman spectroscopy become more widely used to characterize the chemical composition of bone in pre-clinical and translational studies, their bone mineral outcomes require improved validation to inform interpretation of spectroscopic data. In this study, our objectives were (1) to relate Raman and FT-IR carbonate:phosphate ratios calculated through direct integration of peaks to gold-standard analytical measures of carbonate content and underlying subband ratios; (2) to relate Raman and FT-IR MMC measures to gold-standard analytical measures of crystal size in chemical standards and native bone powders. Raman and FT-IR direct integration carbonate:phosphate ratios increased with carbonate content (Raman: p < 0.01, R2 = 0.87; FT-IR: p < 0.01, R2 = 0.96) and Raman was more sensitive to carbonate content than the FT-IR (Raman slope + 95% vs FT-IR slope, p < 0.01). MMC increased with crystal size for both Raman and FT-IR (Raman: p < 0.01, R2 = 0.76; FT-IR p < 0.01, R2 = 0.73) and FT-IR was more sensitive to crystal size than Raman (c-axis length: slope FT-IR MMC + 111% vs Raman MMC, p < 0.01). Additionally, FT-IR but not Raman spectroscopy detected differences in the relationship between MMC and crystal size of carbonated hydroxyapatite (CHA) vs poorly crystalline hydroxyapatites (HA) (slope CHA + 87% vs HA, p < 0.01). Combined, these results contribute to the ability of future studies to elucidate the relationships between carbonate content and fracture and provide insight to the strengths and limitations of FT-IR and Raman spectroscopy of native bone mineral.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Properties of the bone mineral consist of chemical/ compositional factors, such as anionic or cationic substitution, and stoichiometric/ size factors, such as the structural order and crystallite size of the bone mineral. Properties of the bone mineral change with disease, age, and drug treatment and may contribute to bone fragility. For example, bone mineral composition, specifically carbonate content, was a significant explanatory variable between age and BMD matched postmenopausal women with and without fragility fractures. [1,2,3,4] Factors arising from disease, age, and drug treatment such as disrupted cellular remodeling and altered serum chemistry contribute to changes in the properties of bone mineral. Properties of the bone mineral can be quantified with vibrational spectroscopy, energy dispersive X-ray spectroscopy, and nuclear magnetic resonance spectroscopy [5]. Fourier transform infrared (FT-IR) spectroscopy and Raman spectroscopy are the two primary vibrational spectroscopic modalities to rapidly characterize properties of the bone mineral in a spatially resolved fashion. Properties of the bone mineral can be quantified from FT-IR and Raman spectra to measure compositional changes, such as ionic substitution, and structural changes, such as size and stoichiometric perfection [5,6,7]. The two primary FT-IR and Raman outcomes that quantify bone mineral properties are the carbonate:phosphate ratio and mineral maturity/ crystallinity (MMC). The carbonate:phosphate ratio measures the extent of carbonate substitution into the bone mineral crystal lattice, and MMC measures bone mineral crystal size and structural and stoichiometric perfection, with both being related to mechanical properties, aging, bone tissue formation, and remodeling [6, 8,9,10].
Alterations in bone mineral composition (MMC and the carbonate:phosphate ratio) may arise from cellular and physiochemical processes that alter tissue age (time since formation). Mineral maturity/ crystallinity increases with tissue age and animal age and is greater at the center vs. the edge of trabeculae [11,12,13,14]. Mineral maturity/ crystallinity is altered with bone diseases such as osteoporosis, vitamin D deficiency, osteogenesis imperfecta, and renal osteodystrophy [1]. In addition, the carbonate:phosphate ratio was a key explanatory variable for fracture independent of age and BMD in women with fragility fractures, demonstrating its importance as a spectroscopic measure of bone quality [4]. However, there is no well-established relationship between the carbonate:phosphate ratio and tissue age. Carbonate content, typically measured as the carbonate:phosphate ratio, reportedly increases [11] or decreases [12] with tissue age and has greater [15] or lesser [16] values at the edge of trabeculae vs. the center. The inconsistent results on the relationship between the carbonate:phosphate ratio and tissue age could reflect biological variation because bone is compositionally heterogeneous [17]. Variation in aspects of the bone tissue microstructure, such as porosity, contributes to variable interaction of bone mineral with serum sources of carbonate, thereby limiting or promoting carbonate substitution [18]. Furthermore, cellular formation and resorption processes can alter the local chemical environment, disrupting labile mineral surface carbonate content [19, 20]. Therefore, clarification of the underlying physical basis of spectroscopic measures of carbonate substitution can contribute to the understanding of the relationship between the carbonate:phosphate ratio and tissue age.
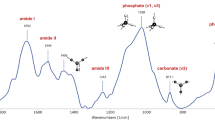
Carbonate is the most common anionic impurity in bone mineral, which is composed of poorly crystalline hydroxyapatite (HA). Carbonate occupies trivalent anionic phosphate sites (B-type), monovalent anionic hydroxide sites (A-type), and unstable labile sites in the hexagonal crystal lattice [21, 22]. The key carbonate bands in bone mineral are ν3 PO4-ν1 CO3 (Raman active), and ν2 CO3 and ν3 CO3 (FT-IR active) (Table 1, Fig. 1 A, B). The Raman ν3 PO4-ν1 CO3 region has been resolved into subbands, and the ν1 CO3-ν3 PO4 component (predominantly ν1 CO3) is correlated with weight percent carbonate [23, 24]. However, the process of spectral deconvolution, second derivative spectroscopy, and subband fitting to resolve these bands is computationally expensive. Furthermore, potential errors between subband fits and pre-processed spectra can occur and are best identified through visual inspection, making these techniques impractical for the analysis of thousands of spectra collected when imaging [25, 26]. Therefore, simple direct integration of the entire ν3 PO4-ν1 CO3 is most commonly used in Raman analysis of mineralized tissues. The FT-IR ν3 CO3: ν1 PO4 ratio has also been related to gold-standard analytical measures of carbonate content, i.e., weight percent carbonate determined by elemental analysis and coulometry [16, 27]. Although the FT-IR ν3 CO3: ν1 PO4 ratio has been validated, it cannot be directly analyzed in native mineralized tissues because of overlapping amide vibrational modes in the ν3 CO3 region (Fig. 1B), making the ν2 CO3 band more commonly used in FT-IR analysis of mineralized tissues. Despite direct integration of the Raman ν3 PO4-ν1 CO3 peak and the FT-IR ν2 CO3 peak being widely used in characterization of mineralized tissue, they have not been related to gold-standard analytical measures of carbonate content. [9, 28,29,30,31,32,33,34]

Characterization of carbonate-substituted hydroxyapatite a Raman spectra and b FT-IR spectra show characteristic phosphate and carbonate vibrational modes, with the carbonate peak size and FWHM of the phosphate peak increasing with carbonate content c Powder XRD diffraction patterns with characteristic (002) and (310) peaks, which decrease in intensity and widen with increasing carbonate content indicating a loss of size and structural perfection
The vibrational spectroscopic outcomes of MMC that have been developed to characterize the poorly crystalline HA in native mineralized tissues are broadly related to crystal size as well as structural and stoichiometric perfection [12, 28]. Specifically, vibrational spectroscopy directly measures mineral maturity as a ratio of apatitic: non-apatitic domains, and indirectly measures crystallinity as a measure of size and degree of structural perfection, by correlations with crystal size assessed by x-ray diffraction (XRD). Notably, mineral maturity and crystallinity, two measures of mineral crystal size and structural and stoichiometric perfection, are highly correlated [35]. Raman and FT-IR MMC metrics are calculated from phosphate peaks in mineralized tissues, which were initially assigned from highly crystalline synthetic HA [13, 23, 36,37,38]. Raman MMC holistically measures particle size and stoichiometric perfection from peak broadening [39]. FT-IR MMC specifically measures local order in the hydroxyapatite crystal lattice as the ratio of subbands in the ν3 PO4 peak at 916–1180 cm−1 (Fig. 1B) corresponding to stoichiometric and non-stoichiometric hydroxyapatite [35, 40]. Raman and FT-IR MMC have been related to the c-axis crystal length, a-axis crystal thickness, carbonate content, and acid phosphate content [14, 40,41,42,43,44,45]. Notably, all previously established correlations between Raman and FT-IR MMC and physical chemical properties have predominantly used chemical standards of highly pure and poorly crystalline HA. However, this system of chemical standards does not reflect large changes in MMC through a range of biologically relevant anionic substitutions, such as carbonate. Therefore, the complex effects of anionic substitution on the relationships between MMC and gold-standard analytical measures of crystal size are unknown.
Despite widespread implementation of vibrational spectroscopic measures to characterize bone mineral, the most frequently used Raman and FT-IR carbonate:phosphate ratios (Raman ν1 CO3: ν1 PO4; FT-IR ν2 CO3: ν3 PO4) are not yet fully validated, and MMC measures remain incompletely understood.[9, 12, 13, 28,29,30, 33, 34, 46,47,48,49,50] Thus, the objectives of this study were to (1) relate Raman and FT-IR carbonate:phosphate ratios calculated through direct integration of peaks to gold-standard analytical measures of carbonate content and underlying subband ratios; (2) to relate Raman and FT-IR mineral maturity/ crystallinity (MMC) measures to gold-standard analytical measures of crystal size in chemical standards of AB-type carbonate-substituted hydroxyapatite (CHA), poorly crystalline hydroxyapatites, and native bone powders.
Methods
Study Design
To broaden the validation of FT-IR and Raman measures of bone mineral carbonate content and crystallinity, we characterized synthetic chemical standards and native bone tissue mineral (Table 2). Synthetic chemical standards enabled modulation of critical variables: (1) carbonate substitution, which was varied over the range 1–14 wt% in carbonate-substituted hydroxyapatite (CHA), and (2) crystal size and stoichiometric perfection, which were varied in poorly crystalline hydroxyapatite, by increasing reaction times in the transition from amorphous calcium phosphate to crystalline hydroxyapatite. Native bone tissue, including deproteinated bone tissue, was characterized to ensure that synthetic hydroxyapatite spanned the biologically relevant range of carbonate substitution, mineral crystal perfection, and mineral crystal size.
To characterize the mineral properties of synthetic chemical standards and native bone mineral, FT-IR spectroscopy, we performed Raman spectroscopy, X-Ray diffraction (XRD), and elemental analysis (Table 2). In all samples, mineral crystal size was characterized with XRD. In carbonate-substituted hydroxyapatite, mineral composition was characterized with FT-IR spectroscopy, Raman spectroscopy, and elemental analysis. The carbonate content of synthetic samples calculated from weight % carbon assessed by elemental analysis is referred to throughout the manuscript as “carbonate content assessed by elemental analysis” because it can be stoichiometrically related to the weight % carbon that is directly measured by this technique. In poorly crystalline HA, mineral composition was characterized with FT-IR and Raman spectroscopy but not elemental analysis because carbonate substitution was not altered in this set of chemical standards. In native bone samples, mineral composition was characterized with FT-IR and Raman spectroscopy but not elemental analysis because the organic phase would contribute to measured carbonate content.
Synthetic Carbonate-Substituted Hydroxyapatite Specimens
Raman and FT-IR carbonate:phosphate ratios and MMC outcomes were measured in chemical standards of AB-type carbonate-substituted hydroxyapatite (CHA) and poorly crystalline hydroxyapatites (HA) synthesized by an aqueous precipitation process [51]. Briefly, 500 mL 10–2 M CaNO3·(H2O)4 was added to 500 mL 5.9 × 10–3 M (NH4)2HPO4 with varying quantities of (NH4)2CO3 while stirring at 65 °C, adjusting the pH to 9.47 ± 0.02 with 28–30% NH4OH. Precipitates were aged in solution for from 0 to 165 h at 65˚C and laundered in 0.15 M NH4OH and 100% ethanol.
Bone Specimens
Bone specimens in this study were a subset of those in a previous study and included as a reference to compare to synthetic chemical standards and ensure that the standards spanned a biologically relevant range of carbonate content from multiple taxa relevant to translational research in osteoporosis treatment [52]. Briefly, specimens included cortical regions of human pyriformis fossa (age = 74, 74, 75, 89 years; sex = F), intracortical regions of murine femoral diaphysis (age = 3 months; sex = F; strain C57Bl/6), and intracortical regions of ovine distal anterior femoral diaphysis (age = 6–8 months; sex = not recorded). Human and murine specimens were collected following procedures approved, respectively, by the Institutional Review Board of the Hospital for Special Surgery and the Institutional Animal Care and Use Committees at Cornell University. The ovine specimens were collected at a slaughterhouse. A subset of the specimens were deproteinated by soaking the specimens in NaOCl for 2 weeks [53]. Specimens were prepared for FT-IR and Raman spectroscopy by dehydration with graded alcohols (70% vol ethanol, 95% vol ethanol, isopropanol, and xylene), de-fatting with isopropyl ether, lyophilizing in a vacuum drier, and powdering in a cryomill (6770 SPEX Sample Prep) [54]. Powder specimens designated for FT-IR spectroscopy (2 mg) were combined with KBr (200 mg) and pressed into pellets with a hydraulic press.
Vibrational Spectroscopy
Raman Spectroscopy
Raman spectroscopy was performed on chemical standards and native bone powders with a Raman microscope (InVia, Renishaw) with a 50X, 0.55 N.A. long-working-distance objective (Lecia Mocrosystems, Vertrieb). Spectra were collected using a 785 cm−1 near-infrared laser at 10mW for 60 s over the range 800–2000 cm−1 with a spectral resolution of 1 cm−1. Raman spectra were analyzed following previously established protocols to generate the second derivative spectra, fit underlying subbands, and calculate the carbonate:phosphate ratio in two ways 1) direct integration of the ν1 CO3-ν3 PO4 peak and 2) with the ν1 CO3-ν3 PO4 subband area, along with the MMC outcome. Prior to analysis, all spectra were baseline corrected to remove fluorescent contributions to the spectra [6, 24, 55, 56].
Direct Integration/ MMC
The computationally efficient direct integration carbonate:phosphate ratio and MMC were calculated from previously established methods using Grams AI (Thermo Fischer Scientific). Briefly, the ν1 PO4 and ν1 CO3-ν3 PO4 peak areas were calculated by linearly baselining the peaks at local minima and integrating them (ν1 PO4 range: 930–980 cm−1; ν3 PO4-ν1 CO3 peak range: 1050–1100 cm−1) [24, 57]. The direct integration Raman carbonate:phosphate ratio was calculated as the area ratio of ν1 CO3-ν3 PO4 / ν1 PO4 peaks, and Raman MMC was calculated as the inverse of the full width at half maximum (FWHM) of the Gaussian distribution fit to the ν1 PO4 peak [41].
Second Derivative Analysis and Subband Peak Fitting
In contrast to the computationally efficient Raman direct integration carbonate:phosphate ratio, relatively computationally expensive second derivative analysis and subband fitting was performed to resolve the ν1 CO3 subband using Grams AI (Thermo Fischer Scientific). Briefly, following truncation over the range 850–1150 cm−1 and smoothing with a 2nd order Savitskay-Golay function, second derivative spectra were calculated and subband peak positions were identified from local minima in the second derivative spectra (Fig. 2a) [58]. Four to seven subbands were fit following previously established methods [24]. Briefly, subbands were input based on the second derivative spectra, and the resulting subband positions, widths, and heights, were determined based on the convergence of the chi-squared error between the fit spectra and the raw spectra using a Levenberg-Marquart algorithm.

Raman spectra of the ν1 CO3 – ν3 PO4 region of synthetic CHA, native human bone, and deproteinated human bone. a Resolution of the Raman spectrum of synthetic CHA (blue) based on the second derivative spectrum (orange) into four distinct subbands (black) fit to the raw spectrum (red); b Resolution of Raman spectrum of human bone (blue) into five distinct subbands (black) fit to the raw spectrum (red dotted line); c Resolution of Raman spectrum of deproteinated human bone (blue) into four distinct subbands (black) fit to the raw spectrum (red dotted line)
FT-IR Spectroscopy
FT-IR spectra of chemical standards and native bone tissue were obtained by dispersing sample powder in KBr at a 100:1 wt/wt KBr:CHA ratio and pressing the powder mixture into pellets. KBr + sample pellets were scanned using a FT-IR spectrometer (Hyperion, Bruker) with a deuterated triglycine sulfate detector from 400 to 2000 cm−1 at a spectral resolution of 2 cm−1. Interferograms were apodized with a medium Norton–Beer function, and FT was performed with two levels of zero filling.
FT-IR Spectral Processing
FT-IR spectra of synthetic CHA and native tissues were analyzed following previously established protocols to calculate the FT-IR carbonate:phosphate ratio, MMC, and acid phosphate content [3, 32, 36, 45]. Spectra were truncated over the range 800–1800 cm−1and baseline-corrected with chemical imaging software (ISYS 5.0, Malvern Instruments, Ltd.). The ν3 PO4 and ν2 CO3 peak areas were calculated by linearly baselining and integrating the peaks (ν3 PO4 range: 916–1180 cm−1; ν2 CO3 range: 852–890 cm−1). The FT-IR carbonate:phosphate ratio was calculated as the area ratio of the ν3 PO4 and ν2 CO3 peaks and the FT-IR MMC was calculated as the intensity ratio at 1030 cm−1/ 1020 cm−1. Acid phosphate content was calculated using the intensity ratio 1127 cm−1/ 1096 cm−1 of the baselined ν3 PO4 peak [32, 36, 45].
Reference Analytical Measures
Elemental Analysis
Elemental analysis was performed on chemical standards by the Cornell University Stable Isotope Laboratory (COIL). The gold-standard analytical measure of carbonate content was determined by C-H-N elemental analysis with an elemental analyzer (NC2500, Carlo Erba) attached to an isotope ratio mass spectrometer (Delta V, Thermo Scientific) calibrated with international reference materials provided by the International Atomic Energy Association. In chemical standards of CHA, weight percent of AB-type carbonate content was calculated by multiplying the weight percent carbon obtained by elemental analysis by a factor of five, the molar mass ratio of carbon/ carbonate. In deproteinated bone, weight percent carbonate was not calculated because the organic phase of bone cannot be completely removed, even after 14 days of deproteinization in NaOCl (Supplemental Fig. S1), consistent with prior studies comparing different methods to deproteinize bone [53]. Thus, bone mineral carbonate content calculated from weight percent carbon obtained from elemental analysis would be inaccurate due to remaining organic carbon sources.
X-Ray Diffraction
Powder X-ray diffraction (XRD) patterns of chemical standards and native tissue were obtained with a powder diffractometer (D8 advance ECO, Bruker) with a 1 kW Cu source and a high-speed silicon strip detector. Three XRD patterns of each sample were obtained at 40 kV and 25 mA: 1) long scans over the range 2θ = 2–45° (15 min scan time) to confirm the hydroxyapatite phase of chemical standards by the absence of a peak at 2θ = 4°, corresponding to octacalcium phosphate (Fig. 1c); 2) short scans over the range 2θ = 22–28° (3 min scan time) to resolve the 002 peak; and 3) short scans from 2θ = 37–43° (3 min scan time) to resolve the 310 peak. Analysis of XRD patterns was performed using powder diffraction software (JADE, Materials Data). The hydroxyapatite phase was confirmed by comparisons of peak profiles to industry standards of synthetic crystalline hydroxyapatite and carbonate-substituted hydroxyapatite (HA JCPDS PDF #98–000-3303, CHA JCPDS PDF #01-072-9861). C-axis crystal length and a-axis crystal thickness were calculated by performing Scherrer analysis on the (002) and (310) short scans, respectively [59].
Statistical Analysis
Statistical analysis was performed using JMP statistical software (SAS) with a significance level of p = 0.05 for all analyses. In synthetic chemical standards, Raman and FT-IR carbonate:phosphate ratios and MMC were correlated to reference analytical measures and subband peak ratios by performing linear regressions, and comparisons of relationships was performed by regression slope analyses by analysis of covariance (ANCOVA).
Results
The presence of distinct peaks in XRD patterns distinguished the CHA phase from the amorphous calcium phosphate (ACP) precursor (Fig. 1c), with the precipitates having a platelet-like morphology 15–40 nm in length determined by Scherrer analysis and confirmed by TEM (Supplemental Fig. S2). Poorly crystalline HAs displayed a transition from ACP, characterized by a lack of distinct peaks in the XRD pattern, to crystalline HA, with characteristic (002) and (310) peaks in the XRD pattern (Supplemental Figure S3 C).
Overall, Raman spectra, FT-IR spectra, and XRD patterns of CHA displayed changes characteristic of increasing AB-type CO3 substitution, as wt% carbonate content was increased in the reaction. Also, Raman spectra, FT-IR spectra, and XRD patterns of HA displayed changes characteristic of increasing structural order during the transition from amorphous calcium phosphate to crystalline HA. In CHA, as we increased reactant sources of carbonate content during synthesis, Raman and FT-IR carbonate peak intensities increased and phosphate peaks widened, and XRD peaks widened (Fig. 1), together suggesting increased AB-type CO3 substitution and decreased crystal size and structural perfection. In poorly crystalline HA, as we increased reaction time, Raman and FT-IR phosphate peak intensities increased and widths narrowed (Supplemental Fig. 3Sa, b). Also, in poorly crystalline HA, distinct peaks in the XRD pattern began to appear with increasing reaction time (Supplemental Figure S3c), suggesting increased crystal size and structural perfection when combined with FT-IR and Raman spectra. These qualitative changes in CHA, poorly crystalline HA, and native bone tissue were reflected in quantitative analysis of peak area ratios, subband area ratios, peak widths, and peak shoulder intensities (see Carbonate:phosphate ratio and Mineral maturity/ crystallinity section).
To assess the validity of our system of chemical standards of CHA, we compared the peak positions of underlying subbands between spectra of CHA and native bone. In spectra of native bone, we identified five peaks corresponding to Raman-active organic and inorganic vibrational modes in the ν1 CO3—ν3 PO4 region of the spectrum (Fig. 2b and Table 3). The positions of these peaks in the spectra of native bone were similar to those in synthetic CHA (Fig. 2a and Table 3).
Carbonate:Phosphate Ratio
All carbonate to phosphate ratios increased with analytical measures of carbonate content (Raman subband: p < 0.01, R2 = 0.92; Raman direct integration: p < 0.01, R2 = 0.87; FT-IR: p < 0.01, R2 = 0.96), and fell within the biological ranges of carbonate:phosphate ratios (Fig. 3a).

a Correlations between FT-IR (maroon) and Raman (green) carbonate:phosphate ratios and gold-standard analytical measures of carbonate content assessed by elemental analysis; b Correlations between 1) Raman subband carbonate:phosphate ratios (green) vs. the Raman imaging direct integration carbonate:phosphate ratio and 2) the FT-IR carbonate:phosphate ratio (maroon) vs. the Raman imaging direct integration carbonate:phosphate ratio. Note, regression equations and Pearson’s squared correlation coefficient are bellow each figure and all p-values for the null hypothesis of the regression slope being zero are p < 0.01
The FT-IR carbonate:phosphate ratio and Raman subband carbonate:phosphate ratio increased with the Raman direct integration carbonate:phosphate ratio (FT-IR: p < 0.01, R2 = 0.93; Raman subband: p < 0.01, R2 = 0.93) (Fig. 3a). Also, the relationship between the FT-IR carbonate:phosphate ratio vs. carbonate content was different than the relationships of both Raman parameters (direct integration and subband) vs. carbonate content (regression slope test: p < 0.01; slope Raman direct integration + 179%, subband + 183% vs. slope FT-IR) (Fig. 3a). However, the relationship between Raman carbonate:phosphate ratios (direct integration and subband) and carbonate content did not differ (regression slope test: p = 0.06).
Mineral Maturity/Crystallinity
Raman and FT-IR MMC correlated with reference analytical measures of crystal size and composition. Specifically, Raman and FT-IR MMC correlated with c-axis crystal length (Raman: p < 0.01, R2 = 0.80; FT-IR p < 0.01, R2 = 0.73) and a-axis crystal thickness (Raman: p < 0.01, R2 = 0.62; FT-IR: p < 0.01, R2 = 0.62). Furthermore, Raman and FT-IR MMC were correlated with each other (p < 0.01, R2 = 0.67). Additionally, a-axis crystal thickness and c-axis crystal length were correlated with each other (p < 0.01, R2 = 0.76). In multivariate regression analysis, c-axis crystal length was a significant explanatory variable of FT-IR and Raman MMC, but a-axis crystal thickness was not a significant explanatory variable (FT-IR MMC: c-axis crystal length p < 0.01, a-axis crystal thickness p = 0.18, R2 = 0.74; Raman MMC: c-axis crystal length p < 0.01, a-axis crystal thickness p = 0.80), suggesting that the carbonate substitutions examined here are volume substitutions in the apatite core that are more affected by c-axis crystal length than substitutions in the hydrated surface layer. Finally, the relationships between Raman MMC and reference analytical measures of crystal size did not differ between CHA and poorly crystalline HAs (regression slope test: c-axis crystal length p = 0.53; a-axis crystal length p = 0.70) but did differ between CHA and poorly crystalline HAs for FT-IR MMC (regression slope test: c-axis crystal length p < 0.01; a-axis crystal length: p = 0.06) (Fig. 4). Specifically, FT-IR MMC was more sensitive to changes in c-axis crystal length in chemical standards consisting of CHA than poorly crystalline HAs (slope CHA + 48% vs. HA) (Fig. 4a).

Correlations between MMC measures assessed by FT-IR (maroon) and Raman (green) spectroscopy with (a, b) crystal length and (c, d) crystal thickness in chemical standards of CHA (solid circles), chemical standards of HA (crossed squares). Native tissue (open circles) were not included in regressions and are provided as a reference to chemical standards. Results of regression analyses of MMC vs. crystal length/thickness for each series of chemical standards (CHA and HA) and Pearson’s squared correlation coefficient are reported beneath each graph (p < 0.01 for all). Vertical lines with p-values between regression equations indicate that the slopes of the pairs of regression lines differed at *p < 0.05 and #p < 0.10 in the regression slope comparison. When slopes of the independent CHA and HA regressions were similar in the regression slope comparison (p > 0.05), as in (b, (c), (d), regression trendlines are plotted for pooled CHA and HA data
In native tissue, FT-IR and Raman measures of MMC and XRD measures of crystal length and thickness were generally clustered in a small range (Fig. 4). Also in native tissue, FT-IR and Raman MMC were systematically below the regression line for chemical standards (Fig. 4). Notably, the length and thickness of crystals in bone specimens were more similar to CHA than to poorly crystalline HA. Mineral crystals from human samples had a wider range of FT-IR MMC but a narrower range of crystal length and thickness than murine and ovine samples (Supplemental Fig. S4 A, B). However, Raman MMC of native bone tissue (human, murine, and ovine) had a narrower range of values than FT-IR MMC (Supplemental Figure S4 C, D), demonstrating that there is an upper limit to which stoichiometry and crystal structure can broaden the Raman ν1 PO4 peak. Finally, deproteinated ovine bone had a wider range of crystal thickness than deproteinated human bone, and a single sample had relatively thin bone mineral crystals (~ 4.4 nm) but did not meet the statistical definition of an outlier (1.5 × interquartile range) (Supplemental Figure S4 C).
Additionally, in chemical standards of CHA, Raman MMC linearly correlated with reference analytical measures of carbonate content, but FT-IR MMC reciprocally correlated with weight % carbonate (Raman linear: p < 0.01, R2 = 0.57; Raman reciprocal: p < 0.01 R2 = 0.34; FT-IR linear: p < 0.01, R2 = 0.58; FT-IR reciprocal p < 0.01, R2 = 0.76) (Fig. 5a, b). Also, Raman and FT-IR MMC correlated with acid phosphate content (Raman: p < 0.01, R2 = 0.52; FT-IR: p < 0.01, R2 = 0.67) (Fig. 5c, d).

Correlations between FT-IR (maroon) and Raman (green) MMC measures with (a, b) gold-standard analytical measures of carbonate content (linear and reciprocal fits are plotted with the fit with the higher Pearson’s squared correlation coefficient (R2) in a bold line and the fit with the lower correlation coefficient in a dotted line) and (c, d) FT-IR acid phosphate content
Discussion
The objectives of this study were to (1) relate computationally efficient Raman and FT-IR carbonate:phosphate ratios calculated through direct integration of peaks to gold-standard analytical measures of carbonate content and underlying subband ratios; (2) to relate Raman and FT-IR mineral maturity/ crystallinity (MMC) measures to gold-standard analytical measures of crystal size in chemical standards of AB-type carbonate-substituted hydroxyapatite (CHA), poorly crystalline hydroxyapatites, and native bone powders. By interrelating Raman and FT-IR measures of carbonate and MMC with their gold-standard analytical counterparts, we provide four key findings that contribute to the understanding and validation of these outcomes used in spectroscopic imaging of native bone tissue. First, all carbonate:phosphate ratios examined (Raman direct integration, Raman subband and FT-IR) were positively correlated with gold-standard analytical measures of carbonate content (Fig. 3a). Second, Raman carbonate:phosphate ratios determined by subband fitting correlated with that determined by direct integration (Fig. 3b) and had similar relationships with gold-standard analytical measures of carbonate content (Fig. 3a). Third, Raman and FT-IR MMC correlated with c-axis crystal length, a-axis crystal thickness, carbonate content, and acid phosphate content (Figs. 4, 5). Fourth, the relationships between FT-IR MMC and reference analytical measures of crystal length differed between CHA and poorly crystalline HAs, but were similar for Raman MMC (Fig. 4a, b ). Additionally, we established that Raman is more sensitive to variations in carbonate content and FT-IR is more sensitive to changes in crystal size and/ or structural perfection, based on quantitative relationships between spectroscopic measures and gold standard analytical measurements. These results are consistent with the fundamental calculation of each outcome: Raman MMC is a general measurement of size and composition from peak broadening, whereas FT-IR MMC is a specific measurement of crystal size and composition from underlying subbands.
Synthetic CHA was structurally changed with incorporation of carbonate into the crystal lattice, with carbonate substitution decreasing the stoichiometric perfection. Distinct peaks in XRD patterns (2θ = 28˚(002) and 2θ = 40˚(310)) (Fig. 1c), FT-IR and Raman spectra (ν3 PO4 and ν1 PO4) confirmed the presence of poorly crystalline synthetic CHA rather than ACP [36, 38]. The presence of CHA was expected because the transition from ACP to HA is expected to occur after 60 min of aging in solution, and the duration of aging in this study was 20 h [41]. Raman and FT-IR carbonate peak intensities increased and phosphate peaks widened with increasing carbonate content (Fig. 1a, b). Also, the νL OH peak at 630 cm−1 was present at low wt% CO3 but was not present at high wt% CO3, corresponding to a loss of hydroxyl groups in the crystal lattice [60]. Hydroxide was initially present in highly pure crystalline hydroxyapatites [HA chemical formula: (Ca5(PO4)6OH)2]. This result suggests that initial carbonate substitution occurred at the A-type (OH−) and B-type (PO42−) sites, causing the νL OH peak to disappear. Then, further predominantly B-type substitution occurred at the PO42− site, forming carbonated HA [CHA, chemical formula: Ca10-2x/3(PO4)6-x(CO3)x(OH)2-x/3]. These results are supported by the previously established inverse relationship between the degree of hydroxylation and atomic order in the crystal lattice [61].
Both Raman carbonate:phosphate ratios (direct integration and subband) are further validated for use in Raman spectroscopy of mineralized tissues by the positive and similar relationships with gold-standard analytical measures of carbonate content (Fig. 3a, b). The positions of the subbands corresponding to Raman-active organic and inorganic vibrational modes in the ν1 CO3—ν3 PO4 region were similar to previously reported values [6, 42]. Furthermore, the calculated subband carbonate:phosphate ratio in CHA fell within the range of native tissue (Fig. 3b), demonstrating the biological relevance of this system of chemical standards. Thus, the Raman direct integration carbonate:phosphate ratio can now be physically related to carbonate content and used for spatially resolved Raman imaging containing thousands of spectra. The FT-IR carbonate:phosphate ratio used to characterize mineralized tissues (ν2 CO3: ν3 PO4 with no overlap between carbonate and amide peaks) was also validated by correlation with gold-standard analytical measures of carbonate content. However, the relationship between the FT-IR carbonate:phosphate ratio and carbonate content was different from those between the Raman carbonate:phosphate ratios and carbonate content (regression slope test: p < 0.01; slope Raman direct integration + 179%, subband + 183% vs. slope FT-IR), indicating FT-IR is less sensitive to carbonate content than its sister modality.
Raman and FT-IR MMC increased with crystal size assessed by XRD. Additionally, in native tissue, FT-IR MMC, Raman MMC, crystal length, and crystal thickness fell within the wide range of values encompassed by CHA and poorly crystalline HA. However, the lower FT-IR and Raman MMC values observed for native tissue compared to those predicted from the regressions for chemical standards (Fig. 4) could potentially arise from additional chemical impurities such as magnesium and fluoride, which can decrease MMC. Differences between human, ovine, and mouse mineral properties could arise from 1) differences in skeletal maturity and microstructure, with the skeletally immature ovine samples composed of rapidly forming plexiform bone with smaller mineral crystals compared to lamellar bone tissue from older humans and 2) murine bones growing through formation/ modeling processes compared to adult humans undergoing cellular remodeling processes. Specifically, Raman and FT-IR MMC increased with c-axis crystal length (Raman: p < 0.01, R2 = 0.80; FT-IR p < 0.01, R2 = 0.73) and a-axis crystal thickness (Raman: p < 0.01, R2 = 0.62; FT-IR: p < 0.01, R2 = 0.62) (Fig. 4), similar to analogous trends observed in different chemical standards [35, 36, 41]. Notably, the correlations between Raman and FT-IR MMC and measures of crystal size were modest (R2 = 0.62–0.80), reflecting the complex nature of MMC, consisting of both crystal size and stoichiometric perfection. Relationships between Raman and FT-IR MMC and XRD crystal size differed by modality (regression slope test: c-axis crystal size: p < 0.01, slope FT-IR MMC + 184% vs. Raman MMC; regression slope test a-axis crystal thickness: p < 0.01, FT-IR MMC + 186% vs. Raman MMC)) (Fig. 4). FT-IR MMC is more sensitive to measures of crystal size than Raman MMC because it is based on subband peak ratios, which have been related to measures of crystal size, whereas Raman MMC is related to crystal size through peak broadening effects [39, 40].
The strengths of the relationships between Raman and FT-IR MMC and measures of crystal size were modest. In particular, they were somewhat weaker than those reported in previous studies due to the interaction between carbonate substitution and crystal size [35, 41]. The variation in MMC vs. crystal size data arises from two parameters: crystal size and crystal stoichiometric perfection, where stoichiometric perfection is influenced by structural order and chemical composition. These two parameters are differentially influenced by carbonate content and crystal maturation time (time since nucleation). In previous studies, a single parameter was modulated (crystal size) by controlling the crystal maturation time while keeping the chemical composition constant, leading to strong correlations between MMC and crystal size [35, 36, 40, 41]. However, in our study, both crystal size and stoichiometric perfection were altered by incorporation of carbonate (CHA) as well as reaction time (HA), leading to modest correlations (R2 = 0.62–0.80). During carbonate incorporation, carbonate ions on the surface of the growing crystal could alter the thermodynamics and kinetics of crystal growth, indirectly altering crystal size. Additionally, A-type and B-type carbonate substitution alter the stoichiometric perfection of the crystal (structural order and chemical composition). Also, incorporation of large amounts of carbonate will increase lattice strain, broadening XRD peaks and thus reducing the calculated crystal size [60]. This lattice strain could further contribute to the modest correlations between Raman and FT-IR MMC and crystal size, especially for larger crystals.
Raman and FT-IR MMC decreased with carbonate content (Fig. 5a ,b). Both Raman and FT-IR MMC appeared to have somewhat non-linear relationships with measures of carbonate content (Fig. 5a, b). This non-linear relationship potentially arises from sequential AB-type and B-type carbonate substitution. The observed two-step process of carbonate substitution (AB-type then B-type) (Fig. 1b) leads to a two-step alteration of MMC. Initially, with low carbonate content, both A-type and B-type substitution occurs. Then, with higher carbonate content, predominantly the more thermodynamically and kinetically favorable B-type substitution occurs. This two-step process suggests that initial carbonate substitution drastically alters the crystal structure perturbing the P-O stretch, and further carbonate substitution only modestly alters the crystal structure. The two-step process we observed is consistent with prior studies that show that carbonate substitution up to 6% wt CO3 drastically alters Raman and P31 NMR peak positions with further substitution only modestly altering peak positions [62]. Also in agreement with prior work, our results suggest that carbonate substitution for phosphate up to one molecule per unit cell functions by a low-ion-loss charge-balance mechanism, with further carbonate substitution functioning by a high-ion-loss mechanism [62].
The observed non-linear relationships between Raman and FT-IR MMC and carbonate content are important for interpreting pre-clinical and translational studies on native bone tissue. FT-IR and Raman MMC and carbonate:phosphate ratios are both associated with tissue age and are therefore relevant to diseases and drug treatments that alter cellular bone remodeling processes. Thus, understanding that carbonate substitution drastically alters MMC in lower quantities, such as when osteoid is initially mineralized, but does not alter MMC as drastically at high quantities, such as in tissue with longer times since mineralization, assists with interpretation of these outcomes. This non-linear relationship may provide insight into potential divergent results between FT-IR and Raman carbonate:phosphate ratios and MMC with tissue age in future studies which characterize actively forming bone tissue, such as at the edge of trabeculae or within osteons, as well as senescent regions of bone, such as at the center of trabeculae and in interstitial regions of the cortex, in groups of pre-clinical and translational interest.
Overall, the strength of the relationship between MMC and crystal size differed by modality. FT-IR MMC was more sensitive to changes in crystal size than Raman MMC (Fig. 4). Additionally, the relationships between Raman MMC and reference analytical measures of crystal size did not differ between CHA and poorly crystalline HAs but did differ between CHA and poorly crystalline HAs for FT-IR MMC (Fig. 4). The different relationships between MMC and crystal length in CHA and HA arise due to the differential mechanisms by which each set of chemical standards alters MMC. In chemical standards of CHA, MMC is predominantly altered through introduction of carbonate impurities into the crystal lattice, introducing lattice strain with modest changes to crystal length. In chemicals standards of poorly crystalline HA, MMC is predominantly altered through crystal growth with modest changes in crystal structure. The ability of FT-IR MMC to differentiate between these two mechanisms, combined with increased sensitivity to crystal size vs. Raman MMC, demonstrate the advantages of FT-IR as a technique to characterize bone mineral. The sensitivity of FT-IR MMC arises because it directly measures both key components of MMC as the ratio of stoichiometric hydroxyapatite (sensitive to crystal size) and non-stoichiometric hydroxyapatite (sensitive to chemical impurities).
The established quantitative relationships between FT-IR and Raman measures of bone mineral composition provide insight into indirect measurements of mechanical performance via FT-IR and Raman spectroscopy. In a recent review, we summarized the relationships between FT-IR and Raman carbonate:phosphate ratios and MMC with mechanical properties [1]. In general, FT-IR and Raman carbonate:phosphate ratios and MMCs correlated with tissue-level measures of indentation properties at the micrometer scale but did not correlate with whole-bone measures of structural properties at the millimeter-centimeter scale. Specifically, several studies have demonstrated that nanoindentation modulus increased with FT-IR and Raman carbonate:phosphate ratios and MMC [8, 63,64,65], although other studies have reported no significant correlations. [50, 66–68] We established two relationships between FTIR and Raman spectroscopic measures of mineral properties and gold-standard analytical measures: 1) carbonate:phosphate ratios and carbonate content and 2) MMCs and crystal size. Therefore, combined with prior work, our relationships suggest that increasing carbonate substitution and mineral crystal size could be associated with increased indentation modulus. However, the mechanical performance of bone arises from a combination of the hierarchical structure and mineral and organic matrix composition. Therefore, compositional assessment alone can only provide modest insight into the mechanical performance of bone at the micrometer scale.
This study has some important limitations and strengths. One limitation is that CHA and HA do not completely replicate the structure and composition of native bone tissue. In addition, acid phosphate content was characterized only with FT-IR data and was not characterized with Raman spectroscopy because resolution of the acid phosphate peak at 1015 cm−1 in the Raman spectrum requires an exceptionally high signal to noise ratio[41], which our instrument did not support, reflecting the inherent challenges with characterizing acid phosphate content in bone. Also, because no gold-standard analytical measurement for acid phosphate content is currently available, this measure itself is incompletely validated. Another limitation of this study is the use of the Raman ν1 PO4 peak, which is moderately polarization dependent [62, 63]. However, because all measurements were made on homogenized, randomly oriented powders, the influence of polarization on ν1 PO4 peak intensity is negligible. A strength of this study is that, for the first time, relationships between computationally efficient Raman and FT-IR carbonate:phosphate ratios and gold-standard analytical measures of carbonate content have been established. In addition, for the first time, Raman and FT-IR measures of MMC have been related to crystal size and composition in a system of chemical standards that incorporates carbonate anionic substitution, the most common anionic substitution in native bone mineral, as well as crystal growth, which occurs in vivo upon mineral nucleation on the osteoid.
The quantitative relationships established in a diverse set of chemical standards and native tissue, combined with prior work [36, 40, 41], contribute to the validation key bone mineral outcomes. These validations allow biomedical researchers to confidently and efficiently characterize the carbonate content of native bone tissue by Raman and FT-IR spectroscopy and identify compositional changes related to disease, age, and drug treatment. Furthermore, this study had the unique capability to identify advantages and disadvantages of each vibrational spectroscopic technique. Overall, FT-IR spectroscopy is more sensitive to changes in bone mineral crystal size and composition than Raman spectroscopy. However, Raman spectroscopy can be used to characterize hydrated tissue, requiring less sample preparation than FT-IR spectroscopy, and is emerging as a new technique for in vivo characterization of bone composition [64, 65]. Thus, these factors should be considered when determining which vibrational spectroscopic modality to choose when performing studies on native bone tissue to improve prediction and understanding of fracture.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Taylor EA, Donnelly E (2020) Raman and Fourier transform infrared imaging for characterization of bone material properties. Bone 139:115490. https://doi.org/10.1016/j.bone.2020.115490
Boskey A, Mendelsohn R (2005) Infrared analysis of bone in health and disease. J Biomed Opt 10:31102. https://doi.org/10.1117/1.1922927
Paschalis EP, Mendelsohn R, Boskey AL (2011) Infrared assessment of bone quality: A review. ClinOrthopRelat Res 469:2170–2178. https://doi.org/10.1007/s11999-010-1751-4
Boskey AL, Donnelly E, Boskey E et al (2016) Examining the relationships between bone tissue composition, compositional heterogeneity, and fragility fracture: a matched case-controlled FTIRI study. J Bone Miner Res 31:1070–1081. https://doi.org/10.1002/jbmr.2759
Hunt HB, Donnelly E (2016) Bone quality assessment techniques: geometric, compositional, and mechanical characterization from macroscale to nanoscale. Clin Rev Bone Miner Metab 14:1–17. https://doi.org/10.1007/s12018-016-9222-4
Mandair GS, Morris MD (2015) Contributions of Raman spectroscopy to the understanding of bone strength. BoneKEy 4:1–8. https://doi.org/10.1038/bonekey.2014.115
Boskey A, Mendelsohn R (2005) Infrared analysis of bone in health and disease. J Biomed Opt 10:031102. https://doi.org/10.1117/1.1922927
Donnelly E, Boskey AL, Baker SP, van der Meulen MCH (2010) Effects of tissue age on bone tissue material composition and nanomechanical properties in the rat cortex. J Biomed Mater Res A 92:1048–1056. https://doi.org/10.1002/jbm.a.32442
Bi X, Patil CA, Lynch CC et al (2011) Raman and mechanical properties correlate at whole bone- and tissue-levels in a genetic mouse model. J Biomech 44:297–303. https://doi.org/10.1016/j.jbiomech.2010.10.009
Lloyd AA, Gludovatz B, Riedel C et al (2017) Atypical fracture with long-term bisphosphonate therapy is associated with altered cortical composition and reduced fracture resistance. ProcNatlAcadSci 114:201704460. https://doi.org/10.1073/pnas.1704460114
Burket J, Gourion-arsiquaud S, Havill LM et al (2011) Microstructure and nanomechanical properties in osteons relate to tissue and animal age. J Biomech 44:277–284. https://doi.org/10.1016/j.jbiomech.2010.10.018
Gourion-Arsiquaud S, Burket JC, Havill LM et al (2009) Spatial variation in osteonal bone properties relative to tissue and animal age. J Bone Miner Res 24:1271–1281. https://doi.org/10.1359/jbmr.090201
Miller LM, Vairavamurthy V, Chance MR et al (2001) In situ analysis of mineral content and crystallinity in bone using infrared micro-spectroscopy of the nu(4) PO(4)(3-) vibration. BiochimBiophysActa 1527:11–19. https://doi.org/10.1016/S0304-4165(01)00093-9
Mendelsohn R, Paschalis EP, Boskey AL (1999) Infrared spectroscopy, microscopy, and microscopic imaging of mineralizing tissues: spectra-structure correlations from human iliac crest biopsies. J Biomed Opt 4:14–21
Boskey AL, DiCarlo E, Paschalis E et al (2005) Comparison of mineral quality and quantity in iliac crest biopsies from high- and low-turnover osteoporosis: An FT-IR microspectroscopic investigation. OsteoporosInt 16:2031–2038. https://doi.org/10.1007/s00198-005-1992-3
Ou-Yang H, Paschalis EP, Mayo WE et al (2001) Infrared microscopic imaging of bone: spatial distribution of CO3(2-). J Bone Miner Res 16:893–900. https://doi.org/10.1359/jbmr.2001.16.5.893
Imbert L, Gourion-Arsiquaud S, Villarreal-Ramirez E et al (2018) Dynamic structure and composition of bone investigated by nanoscale infrared spectroscopy. PLoS ONE 13:1–15. https://doi.org/10.1371/journal.pone.0202833
Cowin SC, Cardoso L (2015) Blood and Interstitial flow in the hierarchical pore space architecture of bone tissue. J Biomech 48:842–854. https://doi.org/10.1016/j.jbiomech.2014.12.013.Blood
Vahidi G, Rux C, Sherk VD, Heveran CM (2020) Lacunar-canalicular bone remodeling: Impacts on bone quality and tools for assessment. Bone. https://doi.org/10.1016/j.bone.2020.115663
Väänänen HK, Laitala-Leinonen T (2008) Osteoclast lineage and function. Arch BiochemBiophys 473:132–138. https://doi.org/10.1016/j.abb.2008.03.037
Rey C, Combes C, Drouet C, Glimcher M (2010) Bone mineral: update on chemical composition and structure. OsteoporosInt 20:1013–1021. https://doi.org/10.1007/s00198-009-0860-y.Bone
Franco W (1994) Mineral, Synthetic and Biological Carbonate Apatites. Stud InorgChem 18:191–304. https://doi.org/10.1016/B978-0-444-81582-8.50009-2
Penel G, Leroy G, Rey C, Bres E (1998) MicroRaman spectral study of the PO4 and CO3 vibrational modes in synthetic and biological apatites. Calcif Tissue Int 63:475–481. https://doi.org/10.1007/s002239900561
Awonusi A, Morris MD, Tecklenburg MMJ (2007) Carbonate assignment and calibration in the Raman spectrum of apatite. Calcif Tissue Int 81:46–52. https://doi.org/10.1007/s00223-007-9034-0
Barth A, Zscherp C (2002) What vibrations tell us about proteins. Q Rev Biophys 35:369–430. https://doi.org/10.1017/S0033583502003815
Baldassarre M, Li C, Eremina N et al (2015) Simultaneous fitting of absorption spectra and their second derivatives for an improved analysis of protein infrared spectra. Molecules 20:12599–12622. https://doi.org/10.3390/molecules200712599
Grunenwald A, Keyser C, Sautereau AM et al (2014) Revisiting carbonate quantification in apatite (bio)minerals: A validated FTIR methodology. J ArchaeolSci 49:134–141. https://doi.org/10.1016/j.jas.2014.05.004
Yerramshetty JS, Lind C, Akkus O (2006) The compositional and physicochemical homogeneity of male femoral cortex increases after the sixth decade. Bone 39:1236–1243. https://doi.org/10.1016/j.bone.2006.06.002
Akkus O, Polyakova-Akkus A, Adar F, Schaffler MB (2003) Aging of microstructural compartments in human compact bone. J Bone Miner Res 18:1012–1019. https://doi.org/10.1359/jbmr.2003.18.6.1012
Kavukcuoglu NB, Arteaga-Solis E, Lee-Arteaga S et al (2007) Nanomechanics and Raman spectroscopy of fibrillin 2 knock-out mouse bones. J Mater Sci 42:8788–8794. https://doi.org/10.1007/s10853-007-1918-x
Donnelly E, Meredith DS, Nguyen JT et al (2012) Reduced cortical bone compositional heterogeneity with bisphosphonate treatment in postmenopausal women with intertrochanteric and subtrochanteric fractures. J Bone Miner Res 27:672–678. https://doi.org/10.1002/jbmr.560
Gourion-Arsiquaud S, Faibish D, Myers E et al (2009) Use of FTIR spectroscopic imaging to identify parameters associated with fragility fracture. J Bone Miner Res 24:1565–1571. https://doi.org/10.1359/JBMR.090414
Schmidt FN, Zimmermann EA, Campbell GM et al (2017) Assessment of collagen quality associated with non-enzymatic cross-links in human bone using Fourier-transform infrared imaging. Bone 97:243–251. https://doi.org/10.1016/j.bone.2017.01.015
Faibish D, Gomes A, Boivin G et al (2005) Infrared imaging of calcified tissue in bone biopsies from adults with osteomalacia. Bone 36:6–12. https://doi.org/10.1016/j.bone.2004.08.019
Farlay D, Panczer G, Rey C et al (2010) Mineral maturity and crystallinity index are distinct characteristics of bone mineral. J Bone Miner Metab 28:433–445. https://doi.org/10.1007/s00774-009-0146-7
Gadaleta SJ, Paschalis EP, Betts F et al (1996) Fourier transform infrared spectroscopy of the solution mediated conversion of amorphous calcium phosphate to hydroxyapatite: new correlations between X-ray diffraction and infrared data. Calcif Tissue Int 58:9–16
Baddiel CB, Berry EE (1966) Spectra structure correlations in hydroxy and fluorapatite. SpectrochimActa 22:1407–1416. https://doi.org/10.1016/0371-1951(66)80133-9
Ou-Yang H, Paschalis EP, Boskey AL, Mendelsohn R (2000) Two-dimensional vibrational correlation spectroscopy of in vitro hydroxyapatite maturation. Biopolym - Biospectroscopy Sect 57:129–139. https://doi.org/10.1002/(SICI)1097-0282(2000)57:3%3c129::AID-BIP1%3e3.0.CO;2-O
Colthup NP, Daly LH, Wiberly SE (1990) Introduction to Infrared and Raman Spectroscopy, 3rd edn. Elsevier Science, New York, NY
Pleshko N, Boskey A, Mendelsohn R (1991) Novel infrared spectroscopic method for the determination of crystallinity of hydroxyapatite minerals. Biophys J 60:786–793
Kazanci M, Fratzl P, Klaushofer K, Paschalis EP (2006) Complementary information on in vitro conversion of amorphous (precursor) calcium phosphate to hydroxyapatite from ramanmicrospectroscopy and wide-angle X-ray scattering. Calcif Tissue Int 79:354–359. https://doi.org/10.1007/s00223-006-0011-9
Morris MD, Mandair GS (2011) Raman assessment of bone quality. ClinOrthopRelat Res 469:2160–2169. https://doi.org/10.1007/s11999-010-1692-y
Paschalis EP, Jacenko O, Olsen B et al (1996) Fourier transform infrared microspectroscopic analysis identifies alterations in mineral properties in bones from mice transgenic for type X collagen. Bone 19:151–156. https://doi.org/10.1016/8756-3282(96)00164-0
Camacho NP, Rinnerthaler S, Paschalis EP et al (1999) Complementary information on bone ultrastructure from scanning small angle X-ray scattering and Fourier-transform infrared microspectroscopy. Bone 25:287–293. https://doi.org/10.1016/S8756-3282(99)00165-9
Spevak L, Flach CR, Hunter T et al (2013) Fourier transform infrared spectroscopic imaging parameters describing acid phosphate substitution in biologic hydroxyapatite. Calcif Tissue Int 92:418–428. https://doi.org/10.1007/s00223-013-9695-9
Freeman JJ, Wopenka B, Silva MJ, Pasteris JD (2001) Raman spectroscopic detection of changes in bioapatite in mouse femora as a function of age and in vitro fluoride treatment. Calcif Tissue Int 68:156–162. https://doi.org/10.1007/s002230001206
Wallace JM, Golcuk K, Morris MD, Kohn DH (2009) Inbred strain-specific response to biglycan deficiency in the cortical bone of C57BL6/129 and C3H/He mice. J Bone Miner Res 24:1002–1012. https://doi.org/10.1359/jbmr.081259
Huang RY, Miller LM, Carlson CS, Chance MR (2002) Characterization of bone mineral composition in the proximal tibia of cynomolgus monkeys: Effect of ovariectomy and nandrolonedecanoate treatment. Bone 30:492–497. https://doi.org/10.1016/S8756-3282(01)00691-3
Donnelly E, Saleh A, Unnanuntana A, Lane JM (2012) Atypical Femoral Fractures Epidemiology, Etiology, and Patient Management. CurrOpin Support Palliat Care. https://doi.org/10.14440/jbm.2015.54.A
Yerramshetty JS, Akkus O (2008) The associations between mineral crystallinity and the mechanical properties of human cortical bone. Bone 42:476–482. https://doi.org/10.1016/j.bone.2007.12.001
Cox SC, Jamshidi P, Grover LM, Mallick KK (2014) Preparation and characterisation of nanophaseSr, Mg, and Zn substituted hydroxyapatite by aqueous precipitation. Mater SciEng C 35:106–114. https://doi.org/10.1016/j.msec.2013.10.015
Taylor EA, Lloyd AA, Salazar-Lara C, Donnelly EL (2017) Raman and FT-IR mineral to matrix ratios correlate with physical chemical properties of model compounds and native bone tissue. ApplSpectrosc. https://doi.org/10.1177/0003702817709286
Su FY, Pang S, Ling YTT et al (2018) Deproteinization of cortical bone: effects of different treatments. Calcif Tissue Int. https://doi.org/10.1007/s00223-018-0453-x
Hunt HB, Torres AM, Palomino PM et al (2019) Altered tissue composition, microarchitecture, and mechanical performance in cancellous bone from men with type 2 diabetes mellitus. J Bone Miner Res 34:1191–1206. https://doi.org/10.1002/jbmr.3711
Nyman JS, Makowski AJ, Patil CA et al (2011) Measuring differences in compositional properties of bone tissue by confocal raman spectroscopy. Calcif Tissue Int 89:111–122. https://doi.org/10.1007/s00223-011-9497-x
Gamsjaeger S, Masic A, Roschger P et al (2010) Cortical bone composition and orientation as a function of animal and tissue age in mice by Raman spectroscopy. Bone 47:392–399. https://doi.org/10.1016/j.bone.2010.04.608
Gamsjaeger S, Buchinger B, Zwettler E et al (2011) Bone material properties in actively bone-forming trabeculae in postmenopausal women with osteoporosis after three years of treatment with once-yearly Zoledronic acid. J Bone Miner Res 26:12–18. https://doi.org/10.1002/jbmr.180
Unal M, Uppuganti S, Leverant CJ et al (2018) Assessing glycation-mediated changes in human cortical bone with Raman spectroscopy. J Biophotonics. https://doi.org/10.1002/jbio.201700352
Cullity B, Stock S (2001) Elements of X-Ray Diffraction, 3rd edn. Prentice Hall, Upper Saddle River, NJ
Venkateswarlu K, Sandhyarani M, Nellaippan TA, Rameshbabu N (2014) Estimation of crystallite size, lattice strain and dislocation density of nanocrystalline carbonate substituted hydroxyapatite by X-ray peak variance analysis. Procedia Mater Sci 5:212–221. https://doi.org/10.1016/j.mspro.2014.07.260
Pasteris JD, Wopenka B, Freeman JJ et al (2004) Lack of OH in nanocrystalline apatite as a function of degree of atomic order: Implications for bone and biomaterials. Biomaterials 25:229–238. https://doi.org/10.1016/S0142-9612(03)00487-3
Kazanci M, Roschger P, Paschalis EP et al (2006) Bone osteonal tissues by Raman spectral mapping: Orientation-composition. J StructBiol 156:489–496. https://doi.org/10.1016/j.jsb.2006.06.011
Kazanci M, Wagner HD, Manjubala NI et al (2007) Raman imaging of two orthogonal planes within cortical bone. Bone 41:456–461. https://doi.org/10.1016/j.bone.2007.04.200
Schulmerich MV, Cole JH, Kreider JM et al (2009) Transcutaneous raman spectroscopy of murine bone in vivo. ApplSpectrosc 63:286–295. https://doi.org/10.1366/000370209787599013
Peterson JR, Okagbare PI, De La Rosa S et al (2013) Early detection of burn induced heterotopic ossification using transcutaneous Raman spectroscopy. Bone 54:28–34. https://doi.org/10.1016/j.bone.2013.01.002
Acknowledgements
The authors thank Phil Carubia and Christopher Umbach for their assistance with collecting FT-IR, XRD, and Raman data; Dr. Michael D. Morris for helpful discussions; Jennie AMR Kunitake for assistance with HA synthesis and for helpful discussions; Dr. Lynn Johnson and the Cornell Statistical Consulting Unit for assistance with data analysis.
Funding
This material is based upon work supported by the National Science Foundation under Grant No. CMMI 1452852. This work made use of the Cornell Center for Materials Research Shared Facilities, which are supported through the NSF MRSEC program (DMR-1719875).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The Authors have no conflicts of interest to disclose.
Research involving Human Participants and/or Animal
Human and murine specimens were collected following procedures approved, respectively, by the Institutional Review Board of the Hospital for Special Surgery and the Institutional Animal Care and Use Committees at Cornell University.
Informed Consent
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Taylor, E.A., Mileti, C.J., Ganesan, S. et al. Measures of Bone Mineral Carbonate Content and Mineral Maturity/Crystallinity for FT-IR and Raman Spectroscopic Imaging Differentially Relate to Physical–Chemical Properties of Carbonate-Substituted Hydroxyapatite. Calcif Tissue Int 109, 77–91 (2021). https://doi.org/10.1007/s00223-021-00825-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-021-00825-4