
Abstract
In the course of chronic kidney disease (CKD), alterations in the bone-vascular axis augment the risk of bone loss, fractures, vascular and soft tissue calcification, left ventricular hypertrophy, renal and myocardial fibrosis, which markedly increase morbidity and mortality rates. A major challenge to improve skeletal and cardiovascular outcomes in CKD patients requires a better understanding of the increasing complex interactions among the main modulators of the bone-vascular axis. Serum parathyroid hormone (PTH), phosphorus (P), calcium (Ca), fibroblast growth factor 23 (FGF23), calcidiol, calcitriol and Klotho are involved in this axis interact with RANK/RANKL/OPG system and the Wnt/β-catenin pathway. The RANK/RANKL/OPG system controls bone remodeling by inducing osteoblast synthesis of RANKL and downregulating OPG production and it is also implicated in vascular calcification. The complexity of this system has recently increased due the discovery of LGR4, a novel RANKL receptor involved in bone formation, but possibly also in vascular calcification. The Wnt/β-catenin pathway plays a key role in bone formation: when this pathway is activated, bone is formed, but when it is inhibited, bone formation is stopped. In the progression of CKD, a downregulation of the Wnt/β-catenin pathway has been described which occurs mainly through the not coincident elevations of sclerostin, Dickkopf1 (Dkk1) and the secreted Frizzled Related Proteins (sFRPs). This review analyzes the interactions of PTH, P, Ca, FGF23, calcidiol, calcitriol and Klotho with the RANKL/RANKL/OPG system and the Wnt/β-catenin, pathway and their implications in bone and cardiovascular disorders in CKD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
General Aspects: Role of Parathormone, Phosphorus and Other Biomarkers
Aging in the general population, which is markedly accelerated in chronic kidney disease (CKD) [1,2,3,4,5,6,7,8], is characterized by severe alterations in the bone-vascular axis that favor osteoporosis, fractures, vascular and soft tissue calcification as well as left ventricular hypertrophy (LVH), myocardial fibrosis and progression of renal damage.
In CKD patients, these abnormalities are associated to elevation in serum parathormone (PTH), phosphorus (P), fibroblast growth factor 23 (FGF23) and decrease in serum calcidiol, calcitriol and calcium (Ca), as well as a progressive reduction in renal Klotho and increase in the degree of systemic inflammation.
In the context of bone and mineral disorders, high serum P stimulates not only PTH synthesis and secretion but also parathyroid gland hyperplasia [9,10,11,12]. These P/PTH interactions create a vicious bone-parathyroid gland circle, high PTH increases bone resorption releasing more P into the circulation and there is a trend to increase serum P levels because CKD patients cannot eliminate P efficiently, not only due to the reduced renal function but also to the decrease in renal Klotho content that impair the phosphaturic response to FGF23.
High PTH induces high bone turnover and bone loss [13], but its effect in the vascular system is still controversial. While PTH 1–34 fragments inhibited vascular calcification in an atherosclerotic murine model [14], PTH 7–84 fragments increased vascular calcification in others [7, 13, 15]. PTH seems to be not sufficient to directly induce vascular calcification [16]. In fact, PTH can have synergistic effects with P, in addition to the well-known capacity of PTH to increase osteoclast activity and bone turnover [16]. A recent study has demonstrated in uremic rats and in cultures of vascular smooth muscle cells (VSMC) that high concentration of PTH increases the calcification induced by high serum P [17]. Furthermore, the silencing of PTH 1 receptor (PTH1R), the most abundant PTH receptor in VSMC, partially abolished the pro-calcifying effect of high PTH, demonstrating an important PTH/PTH1R-driven induction of Ca deposition in the medial artery layer [17].
An additional critical consideration is that in advanced CKD, the serum levels of soluble Klotho cannot reflect the real reduction in renal Klotho content. In fact, soluble Klotho, due to its molecular weight cannot be filtered, so, its appearance in the urine to exert FGF23-independent phosphaturic actions involves a process of transcytosis of soluble Klotho from the blood into the urine through renal tubular cells [18], a process that is impaired in a damaged kidney.
The decrease in urinary soluble Klotho could also partly explain the defects in renal tubular Ca reabsorption and its adverse impact on the skeleton. In fact, urinary soluble Klotho favors the anchoring of the Transient Receptor Potential Cation Channel Subfamily V Member 5 (TRPV5) Ca channel to the cell membrane through its sialidase/glucosidase activity, an action that attenuates the urinary excretion of Ca, preventing a negative Ca balance and, consequently, it could attenuate bone demineralization [19, 20].
This review will analyze the effect and interactions of the above-mentioned factors with the two main bone pathways, Receptor Activator of Nuclear Factor (NF)-ĸB (RANK)/RANK ligand (RANKL)/osteoprotegerin (OPG) and Wnt/β-catenin, describing their implications in bone and cardiovascular disorders in CKD.
The RANK/RANKL/OPG/(LGR4) System
PTH is the main regulator of the RANK/RANKL/OPG system that controls bone remodeling by inducing osteoblast synthesis of RANKL and downregulating OPG production. Both mechanisms favor osteoclastogenesis and bone resorption through a Protein Kinase A (PKA)-driven mechanism [21,22,23]. PKA agonists mimic the PTH regulation of RANKL and OPG gene expression [22, 24].
The OPG/RANK/RANKL system was described in the mid-1990s as an essential regulator of bone modeling and remodeling [25]. Its role in bone maintenance is well known, but some papers attribute to it an important role in vascular calcification [26, 27].
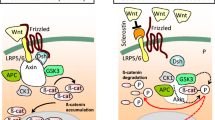
In bone, osteoblasts and osteocytes synthesize and secrete RANKL, which binds to its transmembrane receptor RANK in bone marrow-derived osteoclast progenitors, allowing the maturation, activation and survival of osteoclasts to initiate resorption. In summary, the RANKL action promotes and increases the osteoclastogenesis and bone loss. In addition, osteoblasts secrete OPG, a soluble decoy receptor for RANKL, which prevents the binding of RANKL to RANK, thus attenuating osteoclastogenesis [28] (Fig. 1).

RANK/RANKL/OPG/LGR4 signaling in bone. a Osteoblasts synthesize and secrete RANKL which binds RANK allowing their activation, maturation and prolonging the survival of osteoclasts. b Osteoblasts also secrete OPG, a soluble RANKL decoy receptor, which prevents RANKL binding to RANK, inhibiting osteoclastogenesis. c RANKL can also bind the LGR4 receptor on the surface of the osteoblasts, triggering signals of mineralization and bone formation
RANK, RANKL and OPG: The Classical Components of the Pathway
RANK is fundamental for osteoclast development. It is also called tumor necrosis factor (TNF)-related activation-induced cytokine (TRANCE) receptor and is a member of TNF receptor superfamily. It is a transmembrane receptor that consists of 616 amino acid protein with four extracellular cysteine-rich domains linked to a long C-terminal intracellular region [25, 28,29,30].
RANK is constitutively expressed in multiple organs and cells such us osteoclasts' precursors and mature osteoclasts, dendritic cells, mammary glands and vascular cells, among others. Its functions are associated with bone resorption, immune response, lymph node and mammary gland development and thermal regulation [31,32,33,34,35]. RANK acts as a binder to the cytokine RANKL [31]; however, the RANK overexpression has shown to be enough to activate NF-ĸB [36].
RANKL (also called OPG ligand) is the main stimulator of its specific receptor RANK [36,37,38,39,40]. RANKL is a 316 amino acid protein with a molecular weight of 38 kDa. Its expression is also modulated by several cytokines, glucocorticoids and PTH [41]. RANKL is produced by osteoblasts, osteocytes and activated T cells. It promotes the formation, fusion, differentiation, activation and survival of osteoclasts, allowing increased bone resorption and mineral loss [42, 43] (Fig. 1).
Activation of RANK by RANKL initiates the intracellular signaling cascade of NF-κB (a protein complex that functions as a transcription factor modulating many cellular processes) [44, 45]. Indeed, the final step in RANK activation is the translocation of NF-κB to the nucleus, which can take place through the classical or the alternative route initiated by their respective kinases, named inhibitor of nuclear factor-κB (IκB) kinase (IKK) β and IKKα. The translocation of NF-κB to the nucleus modulates the expression of different genes, such as c-Fos, Nuclear Factor of Activated T cells 1 (NFATc1) and some bone morphogenetic proteins (BMPs) [25, 46].
OPG (also known as osteoclastogenesis inhibitory factor—OCIF) is a decoy receptor for RANKL that regulates osteoclastogenesis disrupting the interaction between RANKL and its receptor RANK [28] (Fig. 1). OPG is a 60 KDa glycoprotein, member of the TNF superfamily that is normally secreted by the osteoblasts, even though it has also been detected in association with the cell membrane in lymphoid cells [47]. OPG consists of 7 structural domains. Domains 1 to 4 give OPG its osteoclastogenesis inhibitory activity, and domains 5 and 6 are considered death domains and involved in apoptosis. Domain 7 harbors the heparin binding region, a common trait of growth factors and signaling molecules [48].
OPG is produced in a wide variety of tissues including the cardiovascular system (heart, arteries and veins), lungs, kidneys, intestine and bone, as well as in hematopoietic and immune cells [30, 47, 49,50,51,52]. Its expression and production are regulated by several cytokines, peptides, hormones and drugs. Cytokines, including TNFα, interleukins 1α and 18, transforming growth factor β (TGFβ), bone morphogenic proteins (BMPs), and steroid hormones, such as 17β estradiol, regulate OPG mRNA levels [53,54,55]. Conversely, glucocorticoids (known to promote bone resorption), immunosuppressant cyclosporin A (which causes osteoporosis and vascular disease), PTH, prostaglandin E2 and fibroblastic growth factor (FGF) decrease OPG expression [53, 56,57,58,59,60].
It is well established that decreases in OPG favor not only increases in osteoclastogenesis and bone resorbing activity but also the increases in vascular Ca deposition. Indeed, OPG reduction was identified as an independent variable for coronary artery calcification [61]. Furthermore, a recent elegant work has linked the anti-calcifying effects of higher vascular Pit2 expression to increases in OPG levels as suggested by the low levels of OPG in the Pit2-deficient mice [62].
In healthy subjects over 70 years old, RANKL and OPG plasma levels were gender-independent [63, 64], but different serum OPG levels were found in men and premenopausal women [65]. There is a discrepancy between OPG concentration and aging, some studies did not observe variations with age [64, 66], while others have shown a positive correlation [63, 65, 67] mainly in subjects over 60 years old [63]. In two age-related disorders, such as rheumatic polymyalgia and osteoarthritis, circulating OPG levels did not differ from the age-matched controls, but soluble RANKL levels were higher in both diseases [63].
The RANK/RANKL/OPG system is of high clinical relevance in osteoporosis treatment. OPG is not used in clinical practice but its discovery and the studies of it actions were the base of the development of Denosumab, a human monoclonal antibody against RANKL that mimics the actions of OPG (reducing osteoclastogenesis and bone resorption). Denosumab has been worldwide long-term used to prevent the reduction of bone mass and to decrease the risk of bone fractures [68].
LGR4: A New RANKL Receptor
The recent discovery of a new RANKL receptor, the leucine-rich repeat-containing G-protein-coupled receptor 4 (LGR4) [69], also called G-protein-coupled receptor (GPR) 48, provided a novel member of this system that regulates bone formation. This receptor counteracts RANKL-driven osteoclastogenesis and also activates the Wnt/β-catenin pathway [70], and it acts on bone formation but may also adversely promote vascular calcification.
LGR4 is essential to increase bone formation by increasing osteoblast maturation and mineralization [69]. In addition, LGR4 inhibits osteoclast differentiation and maturation by the competition with RANK to the binding to RANKL. In this sense, LGR4 knockout mice developed abnormalities in bone during embryonic and postnatal stages with delay in osteoblast differentiation and mineralization, reductions in osteoid formation and increases in osteoclast activity [71]. Also in humans, a nonsense mutation in LGR4 has been strongly associated with low bone mineral density and osteoporotic fractures [72].
However, the effect of PTH on LGR4 expression and the likely mechanisms involved are incompletely understood. A recent study has demonstrated that in uremic rats fed high P diet, LGR4 aortic expression markedly increased in response to high PTH. In vitro, the silencing of the LGR4 gene in VSMCs was capable to prevent PTH-induced vascular calcification without changes in RANKL and OPG expression (73) (Fig. 1). Due to its recent discovery, it is still early to envision if LGR4 will play a future role in the clinical management of bone and vascular disorders.
The RANK/RANKL/OPG/(LGR4) System. Role in Osteoporosis and Vascular Calcification
PTH indirectly regulates osteoclast differentiation and activity by increasing the production of RANKL and decreasing OPG synthesis in osteoblasts [28].
The biological effects of OPG are opposite to those mediated by RANKL, since OPG acts as a soluble inhibitor that prevents the interaction of RANKL with its receptor RANK and, subsequently, its stimulation of osteoclastogenesis [74]. The first evidence that this system was involved in vascular calcification derived from the study of the OPG knockout mouse, which presents osteoporosis and calcification of the aorta and renal arteries [51, 75].
OPG expression can be found in the media of large arteries [51] and in different cell types of the vessel wall such as VSMCs and endothelial cells [58, 76]. In endothelial cells, OPG acts as an autocrine survival factor [76]. In contrast, RANKL and RANK have only been found in the calcified areas of aortas of transgenic mice prone to calcification, but not in the arteries of wild-type mice [77].
The hypothesis that the RANK/RANKL/OPG system could establish a link between osteoporosis and vascular calcification is clinically based on the increased risk of arterial calcifications and cardiovascular disease in postmenopausal women and elderly people with osteoporosis [78,79,80]. Other studies have shown that OPG inhibits the extensive calcifications of the aortic, carotid, femoral, mesenteric, hepatic, renal arteries induced by treatment with warfarin or toxic doses of vitamin D [81]. Moreover, VSMC calcification induced by ß-glycerophosphate or RANKL was inhibited by OPG addition to the culture media [26].
The discovery that the OPG knockout mouse develops osteoporosis and severe arterial calcification [75] and the fact that RANKL expression increases in calcified arterial tissue [82], and also induces calcification of VSMCs through its binding to RANK and increases in BMP 4 expression (26), suggest that the OPG/RANK/RANKL axis could be an important autocrine/paracrine system involved in vascular calcification.
In the vasculature, increases in RANKL and decreases in OPG favor vascular calcification [26, 83]. As it has been previously mentioned, LGR4 seems to promote vascular calcification in experimental models. In addition, LGR4 has also shown to potentiate Wnt/β-catenin pathway through two mechanisms: the increase in Wnt receptors that involve a direct inhibition of the ubiquitinases Ring Finger Protein (RNF) 43 and Zinc And Ring Finger (ZNRF) 3 that mediate their degradation, and also through the recruitment of the guanosine triphosphate (GTP)ase-activating protein Ras GTPase-activating-like protein (IQGAP1) to the Wnt/β-catenin pathway complex, which results in a potentiation of both, the canonical and noncanonical Wnt/β-catenin pathways [84]. Recently, Apelin was identified as an important down-regulator of the activation of LGR4/β-catenin signaling, sufficient to ameliorate aortic remodeling and fibrosis in models of transverse aortic constriction [85]. As for most Wnt inhibitors, beneficial anti-calcifying actions in the vasculature could adversely impact adequate bone formation.
The role of RANKL and OPG as biomarkers of skeletal health has also been studied. In fact, the RANKL/OPG ratio is a useful tool to indirectly determining the degree of bone remodeling [86]; however, its relationship with the degree of vascular calcification [87, 88] is still a controversial issue.
Denosumab has not shown any effect on vascular calcification. In the FREEDOM study (from fracture reduction evaluation of denosumab in osteoporosis every 6 months), performed in osteoporotic women, the abdominal X-rays showed no differences in the progression of aortic calcification and the reported adverse cardiovascular events were found in the placebo and Denosumab groups [89].
WNT/Β-Catenin Pathway
The Wnt/β-catenin is an intracellular signaling pathway that plays a key role in bone formation, regulating the osteoblast activity [90,91,92,93]. When the Wnt/β-catenin pathway is activated, bone is formed, but when this pathway is inhibited, bone formation is stopped.
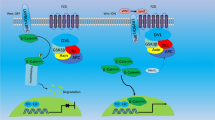
The activation of the Wnt/β-catenin pathway starts when the Wnt ligand binds to their receptors, Frizzled and Low-density lipoprotein receptor-related protein (LRP)5/6, inactivating the Glycogen synthase kinase 3 (GSK3) stabilizing ß-catenin in the cytoplasm making possible the translocation of ß-catenin into the nucleus initiating the transcription of bone forming genes, regulating the preosteoblast differentiation through Runt-related transcription factor 2 (Runx2) or/and Osterix induction, among others [94, 95]. In the absence of the Wnt ligand, ß-catenin is phosphorylated by GSK3, leading to its destruction avoiding its translocation to the nucleus and the osteoblast differentiation and osteocyte formation (Fig. 2).

Wnt/ß-catenin signaling pathway. a In the absence of Wnt ligand, ß-catenin is phosphorylated by GSK3 and destroyed avoiding its translocation to the nucleus to trigger the mechanisms of bone formation. b If Wnt ligands bind to its LRP5/6 and Frizzled co-receptors, GSK3 is inactivated, ß-catenin is stabilized in the cytoplasm and translocate into the nucleus which activates target genes promoting osteoblast differentiation and osteocyte formation. c In presence of Wnt inhibitors, Dkk1 or sclerostin or the sFRPs bind to LRP5/6 receptor or Frizzled, respectively; thus the Wnt/ß-catenin pathway is inhibited. (Modified from Gordon MD et al. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription J Biol Chem. 2006; 281(32): 22,429–22,433 with permission from the American Society for biochemistry and Molecular biology)
The Wnt/β-catenin pathway has inhibitors such us the LRP inhibitors Dickkopf1 (Dkk1) and sclerostin (also called Sost), which bind to LRP5/6 receptor allowing its internalization into the cytoplasm, and the frizzled inhibitors called secreted Frizzled Related Proteins (sFRPs), which are able to block the Wnt/β-catenin pathway, inhibiting an decreasing the osteoblast differentiation and survival, respectively [96,97,98] (Fig. 2).
The Wnt/β-catenin pathway interacts with several hormones and factors such as PTH, FGF23, calcitriol, Klotho and LGR4. The latter is not only a RANKL receptor, but also a key receptor for R-Spondin (R-spo) 2, which is a Wnt/β-catenin pathway activator [99].
PTH is one of the main regulators of Wnt/ β-catenin pathway in bone. It is well known that PTH is an inhibitor of sclerostin, and this action is fundamental to increase bone formation [100,101,102,103], but PTH also affects the differential regulation of LRP5/LRP6 and the antagonist Dkk1 [100]. The use of anti-sclerostin monoclonal antibodies has shown to be effective in preventing bone loss in normal rats and rats with chronic renal failure (CRF) and low serum PTH [104], but not with elevated serum PTH [105], which suggested that serum sclerostin values could be even a more sensitive and precise remodeling marker than circulating PTH. Both continuous and intermittent PTH administration decrease sclerostin levels [102, 103]. Also, continuous PTH increases the signaling receptor Frizzled 1 [103, 106] and co-receptors LRP5 and 6. However, there is no consensus in the effect of PTH over the agonist Dkk1. Several studies have shown that PTH decreases Dkk1 levels [100, 107,108,109] but others have found the opposite effect and PTH treatment was associated with increases in serum Dkk1 levels [110, 111].
Although there is not much evidence about the direct interaction of FGF23/Klotho with Wnt elements, it has been shown that the extracellular domain of Klotho binds to multiple Wnt ligands, inhibiting their ability to activate Wnt signaling [112, 113]. It is interesting to note that in CKD, in parallel to the decrease of Klotho and the increase of FGF23, there are also changes in the levels of Wnt inhibitors, such as sclerostin or Dkk1 [109, 114]. FGF23 directly inhibits the osteoblastic Wnt/β-catenin pathway through a soluble Klotho/mitogen-activated protein kinase (MAPK)-mediated process that requires Dkk1 induction [109]. However, the relationship between FGF23/Klotho and Wnt /β-catenin pathway has not been sufficiently explored.
The soluble Klotho acts as an antagonist of Wnt/β-catenin pathway activation through protein–protein interactions between soluble Klotho and extracellular activators of the Wnt/β-catenin pathway [113]. In CKD, the loss of kidney function is the most important cause of reduction in renal Klotho gene expression. As Klotho downregulates renal calcitriol production, its reduction could influence bone remodeling in CKD patients, acting through the complex PTH-calcitriol-FGF23 axis modulating through a direct protein–protein mechanism, the interaction between the vitamin D receptor (VDR) and β-catenin [115, 116].
Since the activation of the Wnt/β-catenin pathway is also involved in the progression of kidney damage [117], part of the renal and vascular anti-aging effect of blood-soluble Klotho could be explained by its ability to inhibit the Wnt/β-catenin pathway [118, 119]. The interactions between soluble Klotho and the extracellular activators of the Wnt/β-catenin pathway may have negative effects in bone and positive effects in vessels, a matter of current research.
As mentioned earlier, LGR4 (and also LGR5 and LGR6) have been recently identified as second class receptors for the R-spos family and it is another regulator of Wnt/β-catenin pathway [70, 84] through the formation of complexes with recognized Wnt/β-catenin pathway modulators action such us Frizzled/LRP [120]. R-spos activates the Wnt/β-catenin pathway through increasing phosphorylation of the Wnt co-receptors LRP5/6. They cannot directly activate the Wnt/β-catenin pathway, but they can do it indirectly acting as a key receptor for R-spo2, which activates the canonical Wnt/β-catenin pathway promoting osteoblast differentiation and maturation [121,122,123]. A similar effect may occur in osteoblast-like cells derived from VSMCs to initiate vascular calcification. Another member of the family, R-spo1, may also play a role in bone formation by synergizing with the Wnt ligand Wnt3A to induce osteoblast differentiation and OPG expression (99).
The WNT/Β-CATENIN Pathway Inhibitors and Vascular Calcification
As it has been discussed, the Wnt/β-catenin pathway is fundamental for bone formation, but it is also implicated in vascular calcification [17, 124,125,126]. The pathophysiology of vascular calcification involves a transition of the VSMCs to an “osteoblast-like” phenotype; afterwards, a process of mineralization takes place in the vessel [127,128,129]. In this transformation, several hormones and proteins are involved as a promoters or inhibitors of the vascular calcification. It is beyond the scope of this review to list and discuss the role of these factors which have been analyzed in detail in other publications [130,131,132] and also in another review of this special Calcified Tissue International supplement [133]. Among them, the RANK/RANKL/OPG pathway—already discussed in this review—and the Wnt/β-catenin pathway play an important role in bone and vascular metabolism mainly through the inhibitory action of the Wnt/β-catenin pathway through Dkk1, sclerostin and the sFRPs. As a result of the action of these Wnt/β-catenin pathway inhibitors, there is a decrease in the osteoblast differentiation and survival, reducing bone formation [96,97,98] (Fig. 2).
During several decades, the excessive PTH suppression, mainly due to aluminum and Ca load and overdosing of active vitamin D, with the consequent abnormally low serum PTH values, has been considered the main responsible of the pathogenesis of low bone turnover and low bone formation observed in CKD. However, in recent years, this paradigm has been challenged; as a result, the pathogenesis of low bone turnover is under reconstruction and the inhibitors of the Wnt/β-catenin pathway have been blamed as being responsible for the early low bone turnover observed in CKD patients. In fact, clinical and experimental data, both supported by bone biopsy, have shown that bone sclerostin is increased and bone formation decreased in early stages of CKD [17, 109, 134, 135]. Unfortunately, these findings are difficult to translate in the clinical practice to guide therapeutic decisions, due to the fact there is a poor or a lack of correlation between the bone and serum values of the inhibitors of the Wnt/β-catenin pathway, mainly in sclerostin, the most studied molecule [136, 137].
As vascular calcification and bone loss are age-dependent, the association between serum sclerostin levels and age has been investigated. A study performed in healthy pre- and postmenopausal women (aged 20 to 79 years) showed the serum sclerostin significantly increased in postmenopausal women after the menopause [138]. Similarly, another study showed that serum sclerostin levels were 46% higher in old women (mean age, 72.9 years) compared to young women (mean age, 30.0 years), but in contrast, sclerostin mRNA levels measured in bone biopsies were no different in the two groups [139], suggesting the age-dependent decrease in glomerular filtration rate may play a role and it should be considered in the interpretation of serum sclerostin levels.
In experimental models of CRF, sclerostin increases at very early stages, before the increase in P, PTH and FGF23 [109, 134, 140]. Furthermore, studies in humans [134], in a mouse model of slow developing polycystic disease [134, 140] and in a model of CKD with hyperphosphatemia [109] have also shown that the increase in sclerostin in bone precedes the increase in serum P, PTH and FGF23. In fact, the increments of these three factors are a later event which coincides with the decrease in bone sclerostin and with the increments in other inhibitors of the Wnt/β-catenin pathway [109, 134, 140]. In fact, bone biopsies from CKD patients have shown the signals of inhibition in the Wnt/β-catenin pathway were associated with low levels of sclerostin in osteocytes [134], suggesting there may be a contribution of the other inhibitors of the Wnt/β-catenin pathway in the pathogenesis of low bone turnover [141].
In fact, in vitro studies have shown that although the decrease in one Wnt/β-catenin pathway inhibitors is associated with greater vascular calcification, the increase in other Wnt/β-catenin pathway inhibitors may play a role counteracting the decrease and attenuating the effect on vascular calcification [142,143,144]. A good example is a study in diabetic rats with CRF, in which the neutralization of Dkk1 with monoclonal antibodies was sufficient to prevent both bone and vascular damage [145].
Recent studies analyzing the direct effect of PTH and FGF23 on osteoblasts have revealed that elevated PTH inhibits not only the increases in sclerostin, but also the increment of other Wnt/β-catenin pathway inhibitors and that FGF23 may have a direct inhibitory effect on the Wnt/β-catenin pathway in osteoblasts through the induction of Dkk1 [109]. On the contrary, the action of FGF23 would be opposite to that of PTH, since high FGF23, through the induction of increases in Dkk1, would inhibit Wnt/β-catenin pathway in bone contributing to the bone loss, but in vessel could attenuate vascular calcification. In addition to these effects, sclerostin can influence serum concentration of calcitriol and FGF23, both implicated in the mineralization process [146].
The inhibition of sclerostin and other Wnt/β-catenin pathway inhibitors in bone by high levels of PTH could contribute to maintaining bone health, but it is important to highlight that PTH-dependent reduction of the Wnt/β-catenin pathway inhibitors in the vessels could favor vascular calcification. Indeed, as mentioned earlier, recent studies in rats with CKD exposed to different concentrations of PTH suggest that elevated PTH favors vascular calcification. Instead, normal circulating PTH levels appeared to be protective of aortic calcification despite high serum P [17].
It is important to highlight that even though the sclerostin inhibitors, such as Romosozumab, is one of the most promising therapeutic targets in the prevention and treatment of bone fragility fractures [147], its use in CKD patients is still a matter of controversy [148, 149]. In fact, Romosozumab could have a negative action in the vascular system where the “natural” inhibition of the Wnt/β-catenin pathway observed when severe vascular calcification is present can play an important role protecting the vascular wall from further vascular mineralization.
Studies in animals with CKD and severe aortic calcification showed an increased aortic gene expression of some members of the sFRPs family (sFRPs 1, 2 and 4), suggesting the inactivation of Wnt/β-catenin pathway in the vessels wall may constitute a “natural” protective mechanism against the progression of vascular calcification [109, 124]. Thus, according to the present knowledge from experimental models, we can hypothesize that the increase in PTH—a potent sclerostin suppressor—progressively reduces the expression of sclerostin and the increment in other inhibitors, such as Dkk1 and/or sFRPs, of the Wnt/β-catenin pathway could compensate the sclerostin reduction, helping to protect from further vascular calcification [109, 124, 130]. However, another recent study has reported the hypothesis that the increased levels of serum sclerostin probably originating from excessive local production in calcified vessels may contribute to the linkage between vascular disorders and impaired bone mineralization [150].
Cardiac Impact
Among the cardiovascular disorders associated to abnormal Wnt/β-catenin pathway activation, the abnormalities in left ventricular (LV) structure and function are also important. LVH is a well-recognized cardiovascular disorder, which occurs early in the course of CKD [151, 152]. Cardiomyocytes and fibroblasts are the cells implicated in the abnormal remodeling process leading to LVH, the cardiomyocytes increase their size, and the fibroblasts increase collagen synthesis prompting the onset of fibrosis. These changes progressively lead to cardiomyocyte apoptosis or necrosis, and the cardiomyocytes are replaced by fibroblasts and extracellular collagen [153]. In addition, PTH promotes apoptosis of the cardiomyocytes [154], which in the long term either causes or exacerbates myocardial fibrosis.
Several clinical studies have shown that high PTH levels are associated with LVH [155, 156]. Recently, it was demonstrated that myocardial specific R-spo3 acts mainly through the LGR4 receptor to promote coronary stem cell proliferation in the developing heart [157], demonstrating that this receptor and its ligand have an important role in heart development. Moreover, abnormalities in the canonical Wnt/β-catenin pathway are fundamental in the establishment of cardiac lesions. Indeed, activation of β-catenin induces cardiomyocyte hypertrophy and myofibroblastic transformation of cardiac fibroblasts, increasing their ability to produce and secrete interstitial matrix components such as fibronectin and collagen I [158]. Furthermore, a high OPG/RANKL ratio has been independently associated with LVH and abnormal LV structural remodeling in male overweight/obese children and adolescents [159]. A better knowledge of the mechanisms that modulate the appropriate function of the discussed pathways may provide relevant information on novel therapeutic targets to attenuate LVH and myocardial fibrosis in CKD.
FGF23 is also elevated in CKD patients and it has been also related as a cause of LVH [160]. Some authors have speculated that FGF23 could develop LVH through the Wnt signaling activation. In fact, the Wnt signaling inhibition improves cardiac function and could attenuate LV changes [114, 161].
Final Comments
In summary, a better understanding of the intricate regulation of the RANK/RANKL/OPG/LGR4 and the Wnt/β-catenin pathways in bone and vessels is a highly needed step to improve the diagnosis and treatment of these CKD complications.
The design of therapeutic strategies to prevent the deterioration of the bone-vessel axis in the progression of CKD requires a much better understanding of the interaction between classic factors such as Ca, P, calcitriol, PTH and FGF23 with the activation and inactivation of the RANK/RANKL/OPG system and the Wnt/β-catenin pathway. Unfortunately, still it is not possible to translate into the clinical practice a great part of the new important and challenging information discussed in this review because still in many of them, the serum levels of the components of the two main pathways are not able to predict their changes at bone and vascular level.
References
Schulz E, Arfai K, Liu X, Sayre J, Gilsanz V (2004) Aortic calcification and the risk of osteoporosis and fractures. J Clin Endocrinol Metab 89(9):4246–4253
Naves M, Rodriguez-Garcia M, Diaz-Lopez JB, Gomez-Alonso C, Cannata-Andia JB (2008) Progression of vascular calcifications is associated with greater bone loss and increased bone fractures. Osteoporos Int 19(8):1161–1166
Kiel DP, Kauppila LI, Cupples LA, Hannan MT, O’Donnell CJ, Wilson PW (2001) Bone loss and the progression of abdominal aortic calcification over a 25 year period: the Framingham Heart Study. Calcif Tissue Int 68(5):271–276
Frye MA, Melton LJ 3rd, Bryant SC, Fitzpatrick LA, Wahner HW, Schwartz RS et al (1992) Osteoporosis and calcification of the aorta. Bone Miner 19(2):185–194
Cannata-Andia JB, Rodriguez-Garcia M, Carrillo-Lopez N, Naves-Diaz M, Diaz-Lopez B (2006) Vascular calcifications: pathogenesis, management, and impact on clinical outcomes. J Am Soc Nephrol 17(12 Suppl 3):S267–S273
Adragao T, Herberth J, Monier-Faugere MC, Branscum AJ, Ferreira A, Frazao JM et al (2009) Low bone volume–a risk factor for coronary calcifications in hemodialysis patients. Clin J Am Soc Nephrol 4(2):450–455
Coen G, Ballanti P, Mantella D, Manni M, Lippi B, Pierantozzi A et al (2009) Bone turnover, osteopenia and vascular calcifications in hemodialysis patients. A histomorphometric and multislice CT study. Am J Nephrol 29(3):145–152
Chen H, Han X, Cui Y, Ye Y, Purrunsing Y, Wang N (2018) Parathyroid hormone fragments: new targets for the diagnosis and treatment of chronic kidney disease-mineral and bone disorder. Biomed Res Int 2018:9619253
Almaden Y, Canalejo A, Hernandez A, Ballesteros E, Garcia-Navarro S, Torres A et al (1996) Direct effect of phosphorus on PTH secretion from whole rat parathyroid glands in vitro. J Bone Miner Res 11(7):970–976
Almaden Y, Hernandez A, Torregrosa V, Canalejo A, Sabate L, Fernandez Cruz L et al (1998) High phosphate level directly stimulates parathyroid hormone secretion and synthesis by human parathyroid tissue in vitro. J Am Soc Nephrol 9(10):1845–1852
Kilav R, Silver J, Naveh-Many T (1995) Parathyroid hormone gene expression in hypophosphatemic rats. J Clin Invest 96(1):327–333
Slatopolsky E, Brown A, Dusso A (1999) Pathogenesis of secondary hyperparathyroidism. Kidney Int Suppl 73:S14–S19
Neves KR, Graciolli FG, dos Reis LM, Graciolli RG, Neves CL, Magalhaes AO et al (2007) Vascular calcification: contribution of parathyroid hormone in renal failure. Kidney Int 71(12):1262–1270
Shao JS, Cheng SL, Charlton-Kachigian N, Loewy AP, Towler DA (2003) Teriparatide (human parathyroid hormone (1–34)) inhibits osteogenic vascular calcification in diabetic low density lipoprotein receptor-deficient mice. J Biol Chem 278(50):50195–50202
Vattikuti R, Towler DA (2004) Osteogenic regulation of vascular calcification: an early perspective. Am J Physiol 286(5):E686–E696
Graciolli FG, Neves KR, dos Reis LM, Graciolli RG, Noronha IL, Moyses RM et al (2009) Phosphorus overload and PTH induce aortic expression of Runx2 in experimental uraemia. Nephrol Dial Transpl 24(5):1416–1421
Carrillo-Lopez N, Panizo S, Alonso-Montes C, Martinez-Arias L, Avello N, Sosa P et al (2019) High-serum phosphate and parathyroid hormone distinctly regulate bone loss and vascular calcification in experimental chronic kidney disease. Nephrol Dial Transpl 34(6):934–941
Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL et al (2016) Renal production, uptake, and handling of circulating alpha klotho. J Am Soc Nephrol 27(1):79–90
Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG (2005) The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310(5747):490–493
Alexander RT, Woudenberg-Vrenken TE, Buurman J, Dijkman H, van der Eerden BC, van Leeuwen JP et al (2009) Klotho prevents renal calcium loss. J Am Soc Nephrol 20(11):2371–2379
Huang JC, Sakata T, Pfleger LL, Bencsik M, Halloran BP, Bikle DD et al (2004) PTH differentially regulates expression of RANKL and OPG. J Bone Miner Res 19(2):235–244
Fu Q, Jilka RL, Manolagas SC, O’Brien CA (2002) Parathyroid hormone stimulates receptor activator of NFkappa B ligand and inhibits osteoprotegerin expression via protein kinase A activation of cAMP-response element-binding protein. J Biol Chem 277(50):48868–48875
Ben-awadh AN, Delgado-Calle J, Tu X, Kuhlenschmidt K, Allen MR, Plotkin LI et al (2014) Parathyroid hormone receptor signaling induces bone resorption in the adult skeleton by directly regulating the RANKL gene in osteocytes. Endocrinology 155(8):2797–2809
Lee SK, Lorenzo JA (2002) Regulation of receptor activator of nuclear factor-kappa B ligand and osteoprotegerin mRNA expression by parathyroid hormone is predominantly mediated by the protein kinase a pathway in murine bone marrow cultures. Bone 31(1):252–259
Boyce BF, Xing L (2008) Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys 473(2):139–146
Panizo S, Cardus A, Encinas M, Parisi E, Valcheva P, Lopez-Ongil S et al (2009) RANKL increases vascular smooth muscle cell calcification through a RANK-BMP4-dependent pathway. Circ Res 104(9):1041–1048
Kawakami R, Nakagami H, Noma T, Ohmori K, Kohno M, Morishita R (2016) RANKL system in vascular and valve calcification with aging. Inflamm Regen 36:10
Boyce BF, Xing L (2007) Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther 9(Suppl 1):S1
Nelson CA, Warren JT, Wang MW, Teitelbaum SL, Fremont DH (2012) RANKL employs distinct binding modes to engage RANK and the osteoprotegerin decoy receptor. Structure 20(11):1971–1982
Collin-Osdoby P, Rothe L, Anderson F, Nelson M, Maloney W, Osdoby P (2001) Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J Biol Chem 276(23):20659–20672
Sattler AM, Schoppet M, Schaefer JR, Hofbauer LC (2004) Novel aspects on RANK ligand and osteoprotegerin in osteoporosis and vascular disease. Calcif Tissue Int 74(1):103–106
Ono T, Hayashi M, Sasaki F, Nakashima T (2020) RANKL biology: bone metabolism, the immune system, and beyond. Inflamm Regen 40:2
Hanada R, Leibbrandt A, Hanada T, Kitaoka S, Furuyashiki T, Fujihara H et al (2009) Central control of fever and female body temperature by RANKL/RANK. Nature 462(7272):505–509
Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA et al (2000) The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell 103(1):41–50
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T et al (1999) RANK is essential for osteoclast and lymph node development. Genes Dev 13(18):2412–2424
Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER et al (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390(6656):175–179
Green EA, Flavell RA (1999) TRANCE-RANK, a new signal pathway involved in lymphocyte development and T cell activation. J Exp Med 189(7):1017–1020
Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E et al (1999) Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA 96(7):3540–3545
Myers DE, Collier FM, Minkin C, Wang H, Holloway WR, Malakellis M et al (1999) Expression of functional RANK on mature rat and human osteoclasts. FEBS Lett 463(3):295–300
Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Yano K et al (1998) RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun 253(2):395–400
Kong YY, Boyle WJ, Penninger JM (2000) Osteoprotegerin ligand: a regulator of immune responses and bone physiology. Immunol Today 21(10):495–502
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S et al (1999) Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402(6759):304–309
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T et al (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93(2):165–176
Jimi E, Akiyama S, Tsurukai T, Okahashi N, Kobayashi K, Udagawa N et al (1999) Osteoclast differentiation factor acts as a multifunctional regulator in murine osteoclast differentiation and function. J Immunol 163(1):434–442
Wong BR, Besser D, Kim N, Arron JR, Vologodskaia M, Hanafusa H et al (1999) TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell 4(6):1041–1049
Graham TR, Odero-Marah VA, Chung LW, Agrawal KC, Davis R, Abdel-Mageed AB (2009) PI3K/Akt-dependent transcriptional regulation and activation of BMP-2-Smad signaling by NF-kappaB in metastatic prostate cancer cells. Prostate 69(2):168–180
Yun TJ, Chaudhary PM, Shu GL, Frazer JK, Ewings MK, Schwartz SM et al (1998) OPG/FDCR-1, a TNF receptor family member, is expressed in lymphoid cells and is up-regulated by ligating CD40. J Immunol 161(11):6113–6121
Schoppet M, Preissner KT, Hofbauer LC (2002) RANK ligand and osteoprotegerin: paracrine regulators of bone metabolism and vascular function. Arterioscler Thromb Vasc Biol 22(4):549–553
Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Spelsberg TC, Riggs BL (1999) Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology 140(9):4367–4370
Saika M, Inoue D, Kido S, Matsumoto T (2001) 17beta-estradiol stimulates expression of osteoprotegerin by a mouse stromal cell line, ST-2, via estrogen receptor-alpha. Endocrinology 142(6):2205–2212
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R et al (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89(2):309–319
Takai H, Kanematsu M, Yano K, Tsuda E, Higashio K, Ikeda K et al (1998) Transforming growth factor-beta stimulates the production of osteoprotegerin/osteoclastogenesis inhibitory factor by bone marrow stromal cells. J Biol Chem 273(42):27091–27096
Vidal ON, Sjogren K, Eriksson BI, Ljunggren O, Ohlsson C (1998) Osteoprotegerin mRNA is increased by interleukin-1 alpha in the human osteosarcoma cell line MG-63 and in human osteoblast-like cells. Biochem Biophys Res Commun 248(3):696–700
Makiishi-Shimobayashi C, Tsujimura T, Iwasaki T, Yamada N, Sugihara A, Okamura H et al (2001) Interleukin-18 up-regulates osteoprotegerin expression in stromal/osteoblastic cells. Biochem Biophys Res Commun 281(2):361–366
Brandstrom H, Bjorkman T, Ljunggren O (2001) Regulation of osteoprotegerin secretion from primary cultures of human bone marrow stromal cells. Biochem Biophys Res Commun 280(3):831–835
Brandstrom H, Jonsson KB, Ohlsson C, Vidal O, Ljunghall S, Ljunggren O (1998) Regulation of osteoprotegerin mRNA levels by prostaglandin E2 in human bone marrow stroma cells. Biochem Biophys Res Commun 247(2):338–341
Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC et al (1999) Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis1. Endocrinology 140(10):4382–4389
Hofbauer LC, Shui C, Riggs BL, Dunstan CR, Spelsberg TC, O’Brien T et al (2001) Effects of immunosuppressants on receptor activator of NF-kappaB ligand and osteoprotegerin production by human osteoblastic and coronary artery smooth muscle cells. Biochem Biophys Res Commun 280(1):334–339
Nakagawa N, Yasuda H, Yano K, Mochizuki S, Kobayashi N, Fujimoto H et al (1999) Basic fibroblast growth factor induces osteoclast formation by reciprocally regulating the production of osteoclast differentiation factor and osteoclastogenesis inhibitory factor in mouse osteoblastic cells. Biochem Biophys Res Commun 265(1):158–163
Onyia JE, Miles RR, Yang X, Halladay DL, Hale J, Glasebrook A et al (2000) In vivo demonstration that human parathyroid hormone 1–38 inhibits the expression of osteoprotegerin in bone with the kinetics of an immediate early gene. J Bone Miner Res 15(5):863–871
Nitta K, Akiba T, Suzuki K, Uchida K, Watanabe R, Majima K et al (2004) Effects of cyclic intermittent etidronate therapy on coronary artery calcification in patients receiving long-term hemodialysis. Am J Kidney Dis 44(4):680–688
Yamada S, Leaf EM, Chia JJ, Cox TC, Speer MY, Giachelli CM (2018) PiT-2, a type III sodium-dependent phosphate transporter, protects against vascular calcification in mice with chronic kidney disease fed a high-phosphate diet. Kidney Int 94(4):716–727
Pulsatelli L, Dolzani P, Silvestri T, Caraceni P, Facchini A, Ravaglia G et al (2004) Soluble receptor activator of nuclear factor- kappaB Ligand (sRANKL)/osteoprotegerin balance in ageing and age-associated diseases. Biogerontology 5(2):119–127
Jung K, Lein M, Hösslin K, Grosse A, Roth S, Possinger K et al (2002) Osteoprotegerin and receptor activator of nuclear factor-kappaB ligand (RANKL) in the serum of healthy adults. Int J Biol Markers 17(3):177–181
Khosla S, Arrighi HM, Melton LJ 3rd, Atkinson EJ, O’Fallon WM, Dunstan C et al (2002) Correlates of osteoprotegerin levels in women and men. Osteoporos Int 13(5):394–399
Rogers A, Saleh G, Hannon RA, Greenfield D, Eastell R (2002) Circulating estradiol and osteoprotegerin as determinants of bone turnover and bone density in postmenopausal women. J Clin Endocrinol Metab 87(10):4470–4475
Szulc P, Hofbauer LC, Heufelder AE, Roth S, Delmas PD (2001) Osteoprotegerin serum levels in men: correlation with age, estrogen, and testosterone status. J Clin Endocrinol Metab 86(7):3162–3165
Helas S, Goettsch C, Schoppet M, Zeitz U, Hempel U, Morawietz H et al (2009) Inhibition of receptor activator of NF-kappaB ligand by denosumab attenuates vascular calcium deposition in mice. Am J Pathol 175(2):473–478
Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G et al (2016) LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med 22(5):539–546
de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H et al (2011) Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476(7360):293–297
Luo J, Zhou W, Zhou X, Li D, Weng J, Yi Z et al (2009) Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development 136(16):2747–2756
Styrkarsdottir U, Thorleifsson G, Sulem P, Gudbjartsson DF, Sigurdsson A, Jonasdottir A et al (2013) Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature 497(7450):517–520
Carrillo-Lopez N, Martinez-Arias L, Alonso-Montes C, Martin-Carro B, Martin-Virgala J, Ruiz-Ortega M, et al. (2020) The receptor activator of nuclear factor κΒ ligand receptor leucine-rich repeat-containing G-protein-coupled receptor 4 contributes to parathyroid hormone-induced vascular calcification. Nephrol Dial Transpl. https://doi.org/10.1093/ndt/gfaa290
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S et al (1998) Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA 95(7):3597–3602
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C et al (1998) Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12(9):1260–1268
Malyankar UM, Scatena M, Suchland KL, Yun TJ, Clark EA, Giachelli CM (2000) Osteoprotegerin is an alpha vbeta 3-induced, NF-kappa B-dependent survival factor for endothelial cells. J Biol Chem 275(28):20959–20962
Min H, Morony S, Sarosi I, Dunstan CR, Capparelli C, Scully S et al (2000) Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J Exp Med 192(4):463–474
Boukhris R, Becker KL (1972) Calcification of the aorta and osteoporosis. A roentgenographic study. JAMA 219(10):1307–1311
Hak AE, Pols HA, van Hemert AM, Hofman A, Witteman JC (2000) Progression of aortic calcification is associated with metacarpal bone loss during menopause: a population-based longitudinal study. Arterioscler Thromb Vasc Biol 20(8):1926–1931
Kado DM, Browner WS, Blackwell T, Gore R, Cummings SR (2000) Rate of bone loss is associated with mortality in older women: a prospective study. J Bone Miner Res 15(10):1974–1980
Price PA, June HH, Buckley JR, Williamson MK (2001) Osteoprotegerin inhibits artery calcification induced by warfarin and by vitamin D. Arterioscler Thromb Vasc Biol 21(10):1610–1616
Dhore CR, Cleutjens JP, Lutgens E, Cleutjens KB, Geusens PP, Kitslaar PJ et al (2001) Differential expression of bone matrix regulatory proteins in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol 21(12):1998–2003
Weiss RM, Lund DD, Chu Y, Brooks RM, Zimmerman KA, El Accaoui R et al (2013) Osteoprotegerin inhibits aortic valve calcification and preserves valve function in hypercholesterolemic mice. PLoS ONE 8(6):e65201
Carmon KS, Gong X, Lin Q, Thomas A, Liu Q (2011) R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc Natl Acad Sci USA 108(28):11452–11457
Xu R, Zhang ZZ, Chen LJ, Yu HM, Guo SJ, Xu YL et al (2016) Ascending aortic adventitial remodeling and fibrosis are ameliorated with Apelin-13 in rats after TAC via suppression of the miRNA-122 and LGR4-β-catenin signaling. Peptides 86:85–94
Hofbauer LC, Schoppet M (2004) Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA 292(4):490–495
Osorio A, Ortega E, Torres JM, Sanchez P, Ruiz-Requena E (2013) Biochemical markers of vascular calcification in elderly hemodialysis patients. Mol Cell Biochem 374(1–2):21–27
Nitta K, Akiba T, Uchida K, Otsubo S, Takei T, Yumura W et al (2004) Serum osteoprotegerin levels and the extent of vascular calcification in haemodialysis patients. Nephrol Dial Transpl 19(7):1886–1889
Samelson EJ, Miller PD, Christiansen C, Daizadeh NS, Grazette L, Anthony MS et al (2014) RANKL inhibition with denosumab does not influence 3-year progression of aortic calcification or incidence of adverse cardiovascular events in postmenopausal women with osteoporosis and high cardiovascular risk. J Bone Miner Res 29(2):450–457
Komiya Y, Habas R (2008) Wnt signal transduction pathways. Organogenesis 4(2):68–75
MacDonald BT, Tamai K, He X (2009) Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17(1):9–26
Westendorf JJ, Kahler RA, Schroeder TM (2004) Wnt signaling in osteoblasts and bone diseases. Gene 341:19–39
Rawadi G, Roman-Roman S (2005) Wnt signalling pathway: a new target for the treatment of osteoporosis. Expert Opin Ther Targets 9(5):1063–1077
Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD et al (2005) Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA 102(9):3324–3329
Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS et al (2005) Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem 280(39):33132–33140
Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B et al (2003) The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med 349(26):2483–2494
Qiang YW, Barlogie B, Rudikoff S, Shaughnessy JD Jr (2008) Dkk1-induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone 42(4):669–680
Bodine PV, Zhao W, Kharode YP, Bex FJ, Lambert AJ, Goad MB et al (2004) The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol Endocrinol 18(5):1222–1237
Lu W, Kim KA, Liu J, Abo A, Feng X, Cao X et al (2008) R-spondin1 synergizes with Wnt3A in inducing osteoblast differentiation and osteoprotegerin expression. FEBS Lett 582(5):643–650
Kulkarni NH, Halladay DL, Miles RR, Gilbert LM, Frolik CA, Galvin RJ et al (2005) Effects of parathyroid hormone on Wnt signaling pathway in bone. J Cell Biochem 95(6):1178–1190
Silva BC, Bilezikian JP (2015) Parathyroid hormone: anabolic and catabolic actions on the skeleton. Curr Opin Pharmacol 22:41–50
Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA et al (2005) Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146(11):4577–4583
Keller H, Kneissel M (2005) SOST is a target gene for PTH in bone. Bone 37(2):148–158
Moe SM, Chen NX, Newman CL, Organ JM, Kneissel M, Kramer I et al (2015) Anti-sclerostin antibody treatment in a rat model of progressive renal osteodystrophy. J Bone Miner Res 30(3):499–509
Cejka D, Herberth J, Branscum AJ, Fardo DW, Monier-Faugere MC, Diarra D et al (2011) Sclerostin and Dickkopf-1 in renal osteodystrophy. Clin J Am Soc Nephrol 6(4):877–882
Onyia JE, Helvering LM, Gelbert L, Wei T, Huang S, Chen P et al (2005) Molecular profile of catabolic versus anabolic treatment regimens of parathyroid hormone (PTH) in rat bone: an analysis by DNA microarray. J Cell Biochem 95(2):403–418
Guo J, Liu M, Yang D, Bouxsein ML, Saito H, Galvin RJ et al (2010) Suppression of Wnt signaling by Dkk1 attenuates PTH-mediated stromal cell response and new bone formation. Cell Metab 11(2):161–171
Yao GQ, Wu JJ, Troiano N, Insogna K (2011) Targeted overexpression of Dkk1 in osteoblasts reduces bone mass but does not impair the anabolic response to intermittent PTH treatment in mice. J Bone Miner Metab 29(2):141–148
Carrillo-Lopez N, Panizo S, Alonso-Montes C, Roman-Garcia P, Rodriguez I, Martinez-Salgado C et al (2016) Direct inhibition of osteoblastic Wnt pathway by fibroblast growth factor 23 contributes to bone loss in chronic kidney disease. Kidney Int 90(1):77–89
Anastasilakis AD, Polyzos SA, Avramidis A, Toulis KA, Papatheodorou A, Terpos E (2010) The effect of teriparatide on serum Dickkopf-1 levels in postmenopausal women with established osteoporosis. Clin Endocrinol (Oxf) 72(6):752–757
Viapiana O, Fracassi E, Troplini S, Idolazzi L, Rossini M, Adami S et al (2013) Sclerostin and DKK1 in primary hyperparathyroidism. Calcif Tissue Int 92(4):324–329
Wang Y, Sun Z (2009) Current understanding of klotho. Ageing Res Rev 8(1):43–51
Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J et al (2007) Augmented Wnt signaling in a mammalian model of accelerated aging. Science 317(5839):803–806
Muñoz-Castañeda JR, Rodelo-Haad C, Pendon-Ruiz de Mier MV, Martin-Malo A, Santamaria R, Rodriguez M (2020) Klotho/FGF23 and Wnt signaling as important players in the comorbidities associated with chronic kidney disease. Toxins (Basel). 12(3):185
He W, Kang YS, Dai C, Liu Y (2011) Blockade of Wnt/beta-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J Am Soc Nephrol 22(1):90–103
Egan JB, Thompson PA, Vitanov MV, Bartik L, Jacobs ET, Haussler MR et al (2010) Vitamin D receptor ligands, adenomatous polyposis coli, and the vitamin D receptor FokI polymorphism collectively modulate beta-catenin activity in colon cancer cells. Mol Carcinog 49(4):337–352
He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y (2009) Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol 20(4):765–776
Hu MC, Kuro-o M, Moe OW (2013) Renal and extrarenal actions of Klotho. Semin Nephrol 33(2):118–129
Chen T, Mao H, Chen C, Wu L, Wang N, Zhao X et al (2015) The role and mechanism of α-Klotho in the calcification of rat aortic vascular smooth muscle cells. Biomed Res Int 2015:194362
Schuijers J, Clevers H (2012) Adult mammalian stem cells: the role of Wnt, Lgr5 and R-spondins. EMBO J 31(12):2685–2696
Zhu C, Zheng XF, Yang YH, Li B, Wang YR, Jiang SD et al (2016) LGR4 acts as a key receptor for R-spondin 2 to promote osteogenesis through Wnt signaling pathway. Cell Signal 28(8):989–1000
Kim KA, Zhao J, Andarmani S, Kakitani M, Oshima T, Binnerts ME et al (2006) R-Spondin proteins: a novel link to beta-catenin activation. Cell Cycle 5(1):23–26
Nam JS, Turcotte TJ, Smith PF, Choi S, Yoon JK (2006) Mouse cristin/R-spondin family proteins are novel ligands for the Frizzled 8 and LRP6 receptors and activate beta-catenin-dependent gene expression. J Biol Chem 281(19):13247–13257
Roman-Garcia P, Carrillo-Lopez N, Fernandez-Martin JL, Naves-Diaz M, Ruiz-Torres MP, Cannata-Andia JB (2010) High phosphorus diet induces vascular calcification, a related decrease in bone mass and changes in the aortic gene expression. Bone 46(1):121–128
Liao R, Wang L, Li J, Sun S, Xiong Y, Li Y et al (2019) Vascular calcification is associated with Wnt-signaling pathway and blood pressure variability in chronic kidney disease rats. Nephrology (Carlton). https://doi.org/10.1111/nep.13677
Rashdan NA, Sim AM, Cui L, Phadwal K, Roberts FL, Carter R et al (2019) Osteocalcin regulates arterial calcification via altered Wnt signaling and glucose metabolism. J Bone Miner Res. https://doi.org/10.1002/jbmr.3888
Leopold JA (2015) Vascular calcification: mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med 25(4):267–274
Zhu D, Mackenzie NC, Millán JL, Farquharson C, MacRae VE (2011) The appearance and modulation of osteocyte marker expression during calcification of vascular smooth muscle cells. PLoS ONE 6(5):e19595
Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K et al (2000) Phosphate regulation of vascular smooth muscle cell calcification. Circ Res 87(7):E10–E17
Cannata-Andia JB, Roman-Garcia P, Hruska K (2011) The connections between vascular calcification and bone health. Nephrol Dial Transpl 26(11):3429–3436
Cannata Andia J, Carrillo-Lopez N, Rodriguez-Garcia M, Torregrosa JV (2014) Mineral and bone disorders in chronic kidney disease. In: Arici M (ed) Management of chronic kidney disease. Springer, Berlin, pp 223–239
Schlieper G, Schurgers L, Brandenburg V, Reutelingsperger C, Floege J (2016) Vascular calcification in chronic kidney disease: an update. Nephrol Dial Transpl 31(1):31–39
Cannata-Andia J, Martin-Carro B, Martin-Virgala J, Rodriguez-Carrio J, Bande-Fernandez J, Alonso-Montes C et al (2020) Chronic kidney disease—mineral and bone disorders: pathogenesis and management. Calcif Tissue Int. https://doi.org/10.1007/s00223-020-00777-1
Sabbagh Y, Graciolli FG, O’Brien S, Tang W, dos Reis LM, Ryan S et al (2012) Repression of osteocyte Wnt/beta-catenin signaling is an early event in the progression of renal osteodystrophy. J Bone Miner Res 27(8):1757–1772
Massy Z, Drueke T (2017) Adynamic bone disease is a predominant bone pattern in early stages of chronic kidney disease. J Nephrol 30(5):629–634
Behets GJ, Viaene L, Meijers B, Blocki F, Brandenburg VM, Verhulst A et al (2017) Circulating levels of sclerostin but not DKK1 associate with laboratory parameters of CKD-MBD. PLoS ONE 12(5):e0176411
Hansen S, Shanbhogue VV, Jørgensen NR, Beck-Nielsen SS (2019) Elevated bone remodeling markers of CTX and P1NP in addition to sclerostin in patients with X-linked hypophosphatemia: a cross-sectional controlled study. Calcif Tissue Int 104(6):591–598
Ardawi MS, Al-Kadi HA, Rouzi AA, Qari MH (2011) Determinants of serum sclerostin in healthy pre- and postmenopausal women. J Bone Miner Res 26(12):2812–2822
Roforth MM, Fujita K, McGregor UI, Kirmani S, McCready LK, Peterson JM et al (2014) Effects of age on bone mRNA levels of sclerostin and other genes relevant to bone metabolism in humans. Bone 59:1–6
Liu S, Song W, Boulanger JH, Tang W, Sabbagh Y, Kelley B et al (2014) Role of TGF-beta in a mouse model of high turnover renal osteodystrophy. J Bone Miner Res 29(5):1141–1157
Brandenburg VM, Verhulst A, Babler A, D’Haese PC, Evenepoel P, Kaesler N (2019) Sclerostin in chronic kidney disease-mineral bone disorder think first before you block it! Nephrol Dial Transpl 34(3):408–414
Shalhoub V, Shatzen E, Henley C, Boedigheimer M, McNinch J, Manoukian R et al (2006) Calcification inhibitors and Wnt signaling proteins are implicated in bovine artery smooth muscle cell calcification in the presence of phosphate and vitamin D sterols. Calcif Tissue Int 79(6):431–442
Woldt E, Terrand J, Mlih M, Matz RL, Bruban V, Coudane F et al (2012) The nuclear hormone receptor PPARgamma counteracts vascular calcification by inhibiting Wnt5a signalling in vascular smooth muscle cells. Nat Commun 3:1077
Deng D, Diao Z, Han X, Liu W (2016) Secreted frizzled-related protein 5 attenuates high phosphate-induced calcification in vascular smooth muscle cells by inhibiting the Wnt/ss-catenin pathway. Calcif Tissue Int. https://doi.org/10.1007/s00223-016-0117-7
Fang Y, Ginsberg C, Seifert M, Agapova O, Sugatani T, Register TC et al (2014) CKD-induced wingless/integration1 inhibitors and phosphorus cause the CKD-mineral and bone disorder. J Am Soc Nephrol 25(8):1760–1773
Ryan ZC, Ketha H, McNulty MS, McGee-Lawrence M, Craig TA, Grande JP et al (2013) Sclerostin alters serum vitamin D metabolite and fibroblast growth factor 23 concentrations and the urinary excretion of calcium. Proc Natl Acad Sci USA 110(15):6199–6204
McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A et al (2014) Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 370(5):412–420
Evenepoel P, Cunningham J, Ferrari S, Haarhaus M, Kassim Javaid M, Lafage-Proust MH et al (2019) European consensus statement on the diagnosis and management of osteoporosis in chronic kidney disease stages 4 to 5D. Nephrol Dial Transpl. https://doi.org/10.1093/ndt/gfaa192
Evenepoel P, D’Haese P, Brandenburg V (2015) Sclerostin and DKK1: new players in renal bone and vascular disease. Kidney Int 88(2):235–240
De Maré A, Maudsley S, Azmi A, Hendrickx JO, Opdebeeck B, Neven E et al (2019) Sclerostin as regulatory molecule in vascular media calcification and the bone-vascular axis. Toxins (Basel) 11(7):428
McMahon LP, Roger SD, Levin A (2004) Development, prevention, and potential reversal of left ventricular hypertrophy in chronic kidney disease. J Am Soc Nephrol 15(6):1640–1647
Park M, Hsu CY, Li Y, Mishra RK, Keane M, Rosas SE et al (2012) Associations between kidney function and subclinical cardiac abnormalities in CKD. J Am Soc Nephrol 23(10):1725–1734
Cohn JN, Ferrari R, Sharpe N (2000) Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an international forum on cardiac remodeling. J Am Coll Cardiol 35(3):569–582
Fujii H (2018) Association between parathyroid hormone and cardiovascular disease. Ther Apher Dial 22(3):236–241
Acena A, Pello AM, Carda R, Lorenzo O, Gonzalez-Casaus ML, Blanco-Colio LM et al (2016) Parathormone levels are independently associated with the presence of left ventricular hypertrophy in patients with coronary artery disease. J Nutr Health Aging 20(6):659–664
Saleh FN, Schirmer H, Sundsfjord J, Jorde R (2003) Parathyroid hormone and left ventricular hypertrophy. Eur Heart J 24(22):2054–2060
Da Silva F, Massa F, Motamedi FJ, Vidal V, Rocha AS, Gregoire EP et al (2018) Myocardial-specific R-spondin3 drives proliferation of the coronary stems primarily through the Leucine Rich Repeat G Protein coupled receptor LGR4. Dev Biol 441(1):42–51
Zhao Y, Wang C, Hong X, Miao J, Liao Y, Zhou L et al (2018) An essential role for Wnt/beta-catenin signaling in mediating hypertensive heart disease. Sci Rep 8(1):8996
Zampetti S, Lucantoni F, Pacifico L, Campagna G, Versacci P, Pierimarchi P et al (2019) Association of OPG-RANKL ratio with left ventricular hypertrophy and geometric remodeling in male overweight/obese youths. J Endocrinol Invest 42(4):427–434
Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T et al (2011) FGF23 induces left ventricular hypertrophy. J Clin Invest 121(11):4393–4408
Bergmann MW (2010) WNT signaling in adult cardiac hypertrophy and remodeling: lessons learned from cardiac development. Circ Res 107(10):1198–1208
Acknowledgements
The authors wish to thank Instituto de Salud Carlos III (ISCIII; PI17/00715, PI19/00532, PI20/00753), the ISCIII Retic REDinREN (RD06/0016/1013, RD12/0021/0023, RD16/0009/0017 and RD16/0009/0018), Fondo Europeo de Desarrollo Regional (FEDER), Plan Estatal de I + D + I 2013–2016, Plan de Ciencia, Tecnología e Innovación 2013–2017 y 2018–2022 del Principado de Asturias (GRUPIN14-028, IDI-2018–000152), Fundación Renal Iñigo Álvarez de Toledo (FRIAT), and University of Oviedo. N.C.L. has been supported by FINBA-GRUPIN14-028 and IDI-2018-000152, L.M.A. by FINBA-ISCIII (PI17/00384), S.F.V. was supported by FINBA-ISCIII (PI17/00715) and S.P. by FINBA-IDI-2018-000152.
Author information
Authors and Affiliations
Consortia
Contributions
NCL and SP had the idea for the article, LMA and SFV performed the literature search and data analysis, SP, MND and JC drafted the article and NCL, MPRT, AD, JCA, MND and SP critically revised the article.
Corresponding authors
Ethics declarations
Conflict of interest
Natalia Carrillo-López, Laura Martínez-Arias, Sara Fernández-Villabrille, María Piedad Ruiz-Torres, Adriana Dusso, Jorge B. Cannata-Andía, Manuel Naves-Díaz, and Sara Panizo have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The EUROD Workgroup is an initiative of the chronic kidney disease-mineral bone disorder working group of the European Renal Association-European Dialysis and Transplant Association and International Osteoporosis Foundation.
Rights and permissions
About this article

Cite this article
Carrillo-López, N., Martínez-Arias, L., Fernández-Villabrille, S. et al. Role of the RANK/RANKL/OPG and Wnt/β-Catenin Systems in CKD Bone and Cardiovascular Disorders. Calcif Tissue Int 108, 439–451 (2021). https://doi.org/10.1007/s00223-020-00803-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-020-00803-2