
Abstract
Osteoporosis in rheumatic diseases is a very well-known complication. Systemic inflammation results in both generalized and localized bone loss and erosions. Recently, increased knowledge of inflammatory process in rheumatic diseases has resulted in the development of potent inhibitors of the cytokines, the biologic DMARDs. These treatments reduce systemic inflammation and have some effect on the generalized and localized bone loss. Progression of bone erosion was slowed by TNF, IL-6 and IL-1 inhibitors, a JAK inhibitor, a CTLA4 agonist, and rituximab. Effects on bone mineral density varied between the biological DMARDs. Medications that are approved for the treatment of osteoporosis have been evaluated to prevent bone loss in rheumatic disease patients, including denosumab, cathepsin K, bisphosphonates, anti-sclerostin antibodies and parathyroid hormone (hPTH 1–34), and have some efficacy in both the prevention of systemic bone loss and reducing localized bone erosions. This article reviews the effects of biologic DMARDs on bone mass and erosions in patients with rheumatic diseases and trials of anti-osteoporotic medications in animal models and patients with rheumatic diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatic diseases are chronic inflammatory diseases of autoimmune origin. Rheumatoid arthritis (RA) is the most prevalent rheumatic disease with both articular and extra-articular involvements. Bone loss has been recognized as a complication of RA for more than a century [1] and more recently for ankylosing spondylitis (AS) [2, 3]. The bone loss generally presents in two different forms: localized bone erosion with bone loss around an inflamed joint and systemic bone loss, or generalized osteoporosis, which is one of the most common extra-articular manifestations of the disease. The generalized bone loss associated with RA results in an increased risk of osteoporotic fractures in all age groups, both sexes, and various anatomic sites compared to non-RA patients, with an overall relative risk of 2.25 (95% CI 2.25–3.83) [4, 5]. An increased risk of osteoporosis has also been reported for patients with Ankylosing Spondylitis [6].
This paper will discuss the biology of bone loss in RA and studies that have evaluated the effects of medications prescribed for the treatment of rheumatic diseases on localized and generalized bone mass. We refer the readers to the manuscript by Paine and Richlin in this publication [7] for a more detailed description of the biology of bone loss in seronegative spondyloarthropathies, especially psoriatic arthritis (PsA). We will also review studies of localized and generalized bone loss in AS and PsA.
Methods
In order to document the currently available data on both biologic and conventional DMARDs effects on bone turnover and BMD, we performed a primary literature search in PubMed using a few search formulas. We used the following MeSH terms: rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ankylosing spondylitis, hydroxychloroquine, methotrexate, sulfasalazine, leflunomide, TNF-alpha inhibitors, adalimumab, infliximab, certolizumab, golimumab, etanercept, abatacept, CTLA-4 Ig, tocilizumab, rituximab, IL-6 inhibitors, tofacitinib, secukinumab, bone mineral density/BMD, and bone turnover markers. The MeSH terms were searched on PubMed and were limited to articles published in English.
Localized Bone Erosion
In RA, chronic joint inflammation with long standing synovial hyperplasia results in articular bone damage consisting of subchondral cysts, sclerosis, erosive bone damage, and cartilage loss [8, 9]. T cells in the synovium secrete IL-1, IL-6, and IL-17, which induce RANKL production in fibroblast-like synoviocytes (FLS, also known as synovial fibroblasts) [9]. The inflammatory induced increase in RANKL expression is not balanced by an increase in its physiologic inhibitors, mainly osteoprotegerin (OPG) [10]. The binding of RANKL to the RANK receptor on osteoclast precursors and mature osteoclasts leads to the stimulation of several signaling pathways for osteoclast differentiation and activation [9]. In addition, synovial inflammation results in increased production of Wnt/β-catenin signaling proteins, including dickkopf-1 (DKK-1), that inhibit the maturation of osteoblasts [11]. Thus, the interaction between the immune system and skeletal system leads to increased osteoclastogenesis and reduced osteogenesis, which is followed by bone loss.
Systemic Bone Loss
The prevalence of osteoporosis in RA subjects is higher than that observed in an age and gender matched reference group [12]. The risk of fracture is increased at vertebral and appendicular sites of the skeleton [4], and the fracture risk assessment tool FRAX, the most frequently used tool to determine fracture risk worldwide, has RA as one of the seven most important risk factors for fragility fractures [13]. The systemic bone loss in RA is known to involve at least three risk factors: glucocorticoid use, reduced physical activities, and chronic systemic inflammation [14]. The effect of glucocorticoids on bone loss in RA is now interpreted with caution as glucocorticoids have dual effects on bone in RA. Since glucocorticoids reduce the inflammation in RA, they theoretically can reduce some of the localized bone loss [15]; however, glucocorticoids initially increase bone resorption, followed by a delayed and prolonged suppression of bone formation and osteoblast and osteocyte apoptosis [9, 16]. Yao et al. reported glucocorticoid excess in mice resulted in a rapid upregulation of genes involved with osteoclast activation and function, followed by the expression of genes associated with matrix degradation [16]. After a few weeks, the expression of genes associated with osteoblast activation and maturation decreased, and expression of Wingless (Wnt) antagonists, including DKK-1, Wnt inhibitory factor 1, and sclerostin, increased. In addition, there was early increased expression of genes associated with adipogenesis. Mesenchymal stem cells are the precursors of both osteoblasts and adipocytes, and glucocorticoids appear to favor the production of adipocytes [16]. This could lead to a reduction in the number of potential progenitor cells that differentiate into osteoblasts, indirectly affecting bone formation and bone mass.
The pro-inflammatory cytokines that are present in the systemic circulation with inflammatory arthritis stimulate bone resorption, while simultaneously suppressing bone formation. Macrophage colony stimulating factor (M-CSF) and tumor necrosis factor alpha (TNF-α) stimulate the RANK/RANKL pathway [8], and TNF-α is a potent inducer of DKK-1 [17]. The Wnt signaling pathway regulates bone formation via interaction with several signaling pathways, including Wnt/β-catenin pathway (designated as the canonical Wnt pathway), the non-canonical Wnt/planar cell polarity pathway, and the Wnt/calcium pathway [18]. The binding of Wnt ligands to a dual receptor complex, comprising frizzled and either low-density lipoprotein receptor-related protein (LRP) 5 or LRP6, initiates the signaling of the canonical pathway, which leads to inactivation of a multiprotein β-catenin complex. β-Catenin then accumulates in the cytoplasm, and subsequently translocates to the nucleus, where it mediates the transcription of genes that control osteoblast differentiation and OPG, an inhibitor of osteoclast differentiation [19]. The elevated production of DKK-1 by TNF-α reduces Wnt-induced production of OPG, which results in an increase in the RANKL/OPG ratio [17, 18]. The imbalance of RANKL/OPG contributes to accelerated osteoclast resorption, leading to decreased bone mass.
Conventional Synthetic DMARDs, Biologic DMARDs, and Bone Mass in RA
Medications prescribed to reduce joint inflammation for RA and other rheumatic diseases can have effects on bone mass. Conventional synthetic disease modifying agents (DMARDs) including hydroxychloroquine, sulfasalazine (SSZ), methotrexate (MTX), and leflunomide effects on bone mass have been evaluated in a few studies. A study of hydroxychloroquine’s effects on bone turnover in RA patients before and after 3–6 months of treatment, found the level of β-CTx, a marker of bone resorption, at 3 months was not different from the baseline value; however, β-CTx was significantly decreased after 6 months [20]. A cross-sectional study evaluated the effects of SSZ on BMD at the lumbar spine and hip in RA subjects compared to a patient cohort treated with all other available oral DMARDs (MTX, cyclosporin A, chloroquine, injectable gold, auranofin, podophyllin, or penicillamine) and found significantly increased trochanteric BMD with SSZ with no change in the other group [21]. Another study assigned RA patients to treatment with either MTX (15 mg/week) or leflunomide (20 mg/day) for 2.5 years and reported no change in periarticular BMD measured by digital X-ray radiogrammetry or erosions [22].
MTX is the most common DMARD used to treat RA. MTX is frequently used in very high doses to treat malignancies especially in children, and there have been reports of reduced bone mass [23]. A meta-analysis was performed to determine the effect of MTX on bone mass in RA subjects, and of the six studies analyzed, there was no change in femoral neck or lumbar spine BMD for RA or juvenile RA patients on long-term, low-dose MTX [24,25,26,27,28,29,30]. Other studies have evaluated the effects of MTX on bone turnover markers. One study randomly assigned RA subjects to treatment with either salazosulfapyridine (n = 13), actarit (n = 14), or 4–10 mg of MTX (n = 30) per week and assessed study subjects after 3 and 6 months of treatment. There were only changes in the MTX group that included significantly reduced bone resorption marker urine NTX/Cr from the baseline value, and urine DPD/Cr, another resorption marker, also declined; however, it was only significantly different from the baseline value after 6 months, and serum bone alkaline phosphatase (BAP) had no significant change during the 6-month study period [31]. A retrospective study of RA subjects treated with MTX at 7.5 mg/week (n = 30) or SSZ 2 g/day (n = 30) found no significant differences in BMD measured at the lumbar spine and femoral neck between MTX and SSZ groups, or in change in BMD from the baseline value in either group [32]. Studies have also reported fracture risk changes associated with MTX treatment. A cross-sectional and longitudinal study of 117 RA patients over a 4-year period found no change in fracture risk for RA patients taking MTX compared to those not receiving MTX [33].
Tumor Necrosis Factor Alpha Inhibitors
The introduction of biologic DMARDs for the treatment of RA has had a significant influence on both the reduction of inflammation and associated bone loss. TNF-α inhibitors were the first biologic DMARDs to be used in the treatment of RA, and remain the most widely used in clinical practice. At present, there are five TNF-α antagonists approved for the treatment of RA: infliximab, adalimumab, etanercept, certolizumab, and golimumab. In an animal model of collagen-induced arthritis, TNF-α blockage led to a decrease in joint erosions, generalized bone loss, and synovial inflammation [34]. In RA patients, the effect of TNF-α blockade on bone has also been extensively studied. Initial studies of the effect of infliximab on bone turnover markers reported conflicting results. A study of 36 RA patients treated with infliximab and MTX found both the bone resorption marker N-terminal telopeptide (NTX) and the bone formation marker osteocalcin significantly decreased from baseline levels after 14 weeks, while bone resorption marker deoxypyridinoline (DPD) reduction was delayed until 6 months of treatment. Additional follow-up determined no significant changes occurred in any of the bone turnover markers between the 6- and the 12-month measurement, indicating short-term effects on bone turnover in infliximab-treated patients [35]. This observation was supported by a study with 48 infliximab-treated RA patients that showed decreased levels of CTX-1 at 6 and 22 weeks after biologic administration, but levels returned to baseline at 54 weeks [36]. However, another study of 68 RA patients treated with infliximab found after 6 weeks of treatment, there was an increase in the bone formation markers osteocalcin and N-propeptide of type I procollagen (P1NP) from 21.2 to 23.9 ng/mL and 43.9 to 50.1 µg/mL respectively, and a decrease in the bone resorption marker C-terminal cross-linking telopeptide of type I collagen generated by matrix metalloproteinases (ICTP) from 8.9 to 7.8 µg/mL [37]. Another study of 90 RA subjects treated with infliximab for 1-year reported no change in the serum level of osteocalcin and the bone resorption marker CTX-1 [38]. In addition, 102 infliximab-treated RA patients had significant reductions in the resorption marker B-CTX at 14, 30, and 46 weeks [39]. Finally, Lange et al. reported data on 26 RA patients treated with infliximab for 1 year, and found a significant increase in osteocalcin and a significant decrease in CTX-1 [40]. In summary, changes in markers of bone formation and resorption are present with TNF blocking agents. Also, the baseline bone turnover of the RA patients at the time of the initiation of the biologic appears to influence the changes induced by the biologic and bone resorption generally declines.
The introduction of TNF inhibition for the treatment of RA patients has been associated with decreased generalized bone loss [17]. In an open label study, 50 RA patients treated for 1 year with adalimumab + MTX, with or without less than 10 mg/day of prednisone, had no reduction in BMD of the lumbar spine or femur [41]. Another open label study treated 36 RA patients with infliximab for 1 year, measured lumbar spine and hip BMD, and a found a non-significant decrease in hip and increase in lumbar spine BMD [42]. When comparing 90 RA infliximab-treated patients after 1 year of treatment to retrospective data on 99 MTX-treated patients, there was less BMD decrease in the infliximab-treated cohort at lumbar spine and hip [38]. A larger study, the BeST study, randomized 342 RA patients to receive (1) sequential monotherapy (MTX, SSZ, then leflunomide), (2) step-up combination therapy (MTX, SSZ, then hydroxychloroquine), (3) initial combined therapy with glucocorticoids (MTX or SSZ with prednisone), and (4) initial combined therapy with infliximab (MTX with infliximab). At 1-year follow-up, median lumbar spine and hip BMD decreased by 0.8 and 1%, and showed no variation between groups [43].
Multiple other studies reported on both central and localized bone changes. An open cohort study on 102 RA patients treated with infliximab for 1 year reported no reduction in BMD of lumbar spine and hip; however, there was progressive metacarpal cortical bone loss of 0.8% [39]. Also, 52 RA patients treated for more than 2 years with infliximab had BMD measurements at baseline, and after 2 years and had no change in the BMD of the lumbar spine and hip, while bone mass of the metacarpal bones declined [44]. In another cohort study consisting of 184 RA patients treated with adalimumab for 1 year, there was no change in BMD at lumbar spine and hip, but metatarsal BMD was significantly decreased by 1.41%. The study also included a follow-up after 4 years of treatment and found a significant decrease in BMD measured at the hip, but no change in BMD measured at the lumbar spine. The metatarsal bone loss continued to decrease by an additional 0.83%/year [45].
IL-6 Inhibition
Interleukin-6 (IL-6) inhibitors approved for the treatment of RA are known to inhibit osteoclast activity, which may over time influence local and generalized bone mass in RA subjects. The first approved IL-6 inhibitor, tocilizumab, effects on bone turnover and BMD have been studied, and results vary. The OPTION study was the first to analyze tocilizumab effects on bone turnover markers. This multicenter, double-blind, placebo-controlled trial included 416 patients, analyzed the bone resorption markers CTX-1 and ICTP, and found a significant decrease in both bone resorbing markers at 4, 16, and 24 weeks, and an increase in the bone formation marker PINP, which was only significant 4 weeks after treatment initiation [46]. A second large, randomized, double-blind, placebo-controlled, trial analyzed 299 patients treated with either tocilizumab + MTX, or placebo + MTX and found a significant reduction in the CTX-1/osteocalcin ratio at 24 weeks [47]. Briot et al. studied RA patients, treated with standard DMARDs for RA and 8 mg/kg of tocilizumab for 44 weeks, and found no change in lumbar spine or total hip BMD, while Chen et al. treated RA patients with tocilizumab for 2 years and reported an increase in femoral neck BMD, but a non-significant increase in lumbar spine BMD for a subgroup of anti-citrullinated protein antibody (ACPA) positive patients. However, there was no change in the subgroup of RA patients negative for ACPA [48, 49]. Both studies also measured bone formation using P1NP and bone breakdown using CTX, and Chen et al. also measured osteocalcin. While Briot et al. reported an increase in P1NP and no change in CTX, Chen et al. reported a decrease in CTX and no change in P1NP or osteocalcin. An additional study measured BMD at the lumbar spine and femoral neck 52 weeks after initiating tocilizumab treatment, and found no change in BMD at either the lumbar spine or femoral neck for patients without osteopenia, but a significant increase in both locations for patients with baseline osteopenia [50]. In summary, tocilizumab appears to reduce the increased bone turnover observed in inflammatory arthritis and to improve bone mass in subgroups of RA patients that are ACPA positive, with low bone mass when initiating therapy and treated for 2 years.
Currently, information on bone turnover and mass changes for Kevzara, another IL-6 antagonist recently approved, are not yet available.
Janus Kinase 1 and 3 Inhibitor
Tofacitinib is a selective inhibitor of Janus kinase 1 (JAK1) and Janus Kinase 3 (JAK3) that interferes with the dimerization of signal transducers and activation of transcription (STAT) molecules. This results in inhibition of STAT1 and STAT3 of the JAK-STAT signaling pathway [51]. The efficacy and safety of tofacitinib in active moderate-to-severe RA has been demonstrated, and the inhibition of structural damage in patients receiving tofacitinib has been shown using radiographs [52, 53]. Clinical studies evaluating local and generalized bone mass have not yet been reported with tofacitinib. However, a recent study evaluated the bone effects of tofacitinib in the adjuvant-induced arthritis rat model [54]. Tofacitinib treatment did not prevent inflammation-induced loss of cortical and trabecular bone structure and strength, although at the tissue level bone hardness, measured by nanoindentation, was conserved compared to placebo controls. The difference between local and systemic bone effects of tofacitinib could be explained by complex interactions with bone-related cells by intracellular molecules [54]. JAK1 is expressed in bone cells and is involved in bone formation. Thus, depletion of JAK1 may cause a delay in bone growth. STAT1 inhibits transcription of Runx2 in osteoblasts, and the inactivation of STAT1 leads to an osteoporotic bone phenotype in both Chinese and Caucasians [55]. The JAK-STAT3 signal transduction pathway promotes osteoblastic differentiation. Inactivation of STAT3 in osteoblasts led to lower bone mass and STAT3 mutations increased osteoclast number, bone resorption, and was associated with recurrent fractures in mice [56]. Clinical studies are now underway to determine the long-term effects of tofacitinib on localized and generalized bone mass in rheumatic disease patients.
Anti-CD20 Depletion Rituximab
Rituximab is a CD20 B cell depleting antibody that is approved for the treatment of RA after an ineffective response to oral DMARDs or other biologic agents. To evaluate the effects of rituximab on bone metabolism, a prospective study consisting of 13 patients treated with rituximab measured serum bone turnover markers including alkaline phosphatase, CTX, tartrate-resistant acid phosphatase, and DPD 15 months after treatment was initiated. No changes were found in bone formation markers or tartrate-resistant acid phosphatase, but there was a significant decrease in DPD [57], suggesting a reduction in osteoclast activity. Another open label study measured osteocalcin and NTX in 28 patients before and after 16 weeks of rituximab treatment. There was a non-significant increase in osteocalcin, and no change in NTX [58]. Finally, 16 patients with RA were treated with rituximab for 18 months and subjects classified as clinical responders had an improvement in lumbar spine BMD, and no change in femoral BMD, while in non-responders, there was an improvement in lumbar spine BMD, but a decrease in femoral BMD [59].
Anti-CTLA-4Ig Abatacept
Abatacept is fusion protein composed of the Fc region of the immunoglobulin IgG1 fused to the extracellular domain of CTLA-4 that is approved for the treatment of RA after an ineffective response to other DMARDs or biologic agents. A murine model of inflammatory arthritis showed that CTLA-4Ig inhibited differentiation and maturation of osteoclasts, suggesting that this antibody may have anti-resorptive effects. This observation was confirmed in an in vitro study, as the addition of CTLA-4Ig dose-dependently decreased osteoclast formation and osteoclast activity measured by a resorption assay [60]. One study in humans randomized 165 RA patients to abatacept or a treatment group receiving either infliximab, etanercept, adalimumab, tocilizumab, or golimumab, and measured BMD before and after 1 year of treatment. They found no significant difference in BMD measured at the lumbar spine, but found a significantly higher increase in BMD measured at the femoral neck in the abatacept treatment group compared to the group receiving other treatments [61].
Ankylosing Spondylitis (AS)
AS is a chronic systemic inflammatory disease that affects the spine and sacroiliac joints [62]. Osteoporosis is considered a common feature of AS, even in early stages of the disease [63]. The prevalence of osteoporosis was 25% at mean age of 38.9 years in AS patients according to the WHO classification, and vertebral fracture was detected in 43%. In a clinical series of 111 AS patients, 23 patients had either a compression or biconcave spinal fracture [3].
The skeletal phenotype of seronegative spondyloarthropathies (e.g., AS and Psoriatic Arthritis, PsA) is different from that observed in RA, as in the former there can be pathologic bone formation. This new bone formation may present as syndesmophytes that are usually symmetric and can be bridging in AS, sacroiliitis, or sacroiliac joint ankylosis. Bone formation in the peripheral skeleton may present with enthesophytes, periosteal new bone formation, and ossification of the enthesis.
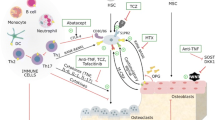
The biologic bone mass changes in the axial and peripheral skeleton in AS differ from RA. In AS, there is increased bone resorption in the axial and peripheral skeleton due to systemic inflammation, while there can be an increase in bone formation at the enthesis sites. Recent studies from the mouse enthesis have identified T cells with an IL23 receptor, which when stimulated with IL-23 increase the production of other pro-inflammatory cytokines including TNF, interleukin-17 (Il-17), and interleukin-22 [7, 64, 65]. Also IL-23 can increase the production of bone morphogenetic proteins that can stimulate new bone formation. Overexpression of the IL-23 in mice produced enthesitis and arthritis with features like SpA [66]. These new observations help to explain how our anti-rheumatic treatments help patients with AS (Fig. 1).

Site of action of biologic anti-rheumatic medications and osteoporosis medications
Conventional Oral DMARDs
Conventional oral DMARDs, including sulfasalazine and MTX, have not been studied for their effects on localized and generalized bone loss in AS subjects.
Biologic Agents and BMD in AS
TNF blockers have been successfully used to reduce disease activity in the treatment of ankylosing spondylitis [67]. Infliximab was shown to increase BMD in 29 AS patients by 3.6% at the lumbar spine and 2.2% at the total hip, compared to baseline after 6 months [68]. In support of these results, when 279 patients were randomly assigned to placebo or infliximab, the infliximab group showed significantly greater increased BMD measured at the spine and hip than placebo at 24 weeks [69]. Another study assessed BMD in 90 AS patients in four different treatment groups: (1) conventional treatment (NSAID, SSZ, or MTX), (2) bisphosphonate + conventional, (3) anti-TNF agent (infliximab, etanercept, and adalimumab) + conventional, or (4) bisphosphonate + Anti-TNF agent + conventional. At follow-up of less than 3 years, there was a significant increase in lumbar spine BMD for all groups except bisphosphonate + conventional treatment. There was no significant change in BMD measured at femoral neck, upper neck, or trochanter for any groups [70]. The increased BMD are likely confounded by the spine ossification, a chronic post-inflammatory feature that is characteristic of AS, because these ossifications usually require many years to develop, and the effect of the biologics on BMD in AS is usually fairly rapid [20]. Another study evaluated bone formation in 29 patients with SpA unresponsive to MTX, SSZ, or NSAIDs after 6 months of infliximab treatment, and found the bone formation marker osteocalcin to be elevated with the median change of 1.45 µg/L (− 2.10 to 42.10, p = 0.013) compared to the baseline value [68]. Radiographs of the cervical and lumbar spine from 257 AS patients treated with etanercept for up to 96 weeks were compared with radiographs from 175 AS patients in a large prevalence cohort who had not been treated with anti-TNF agents, and there was no structural benefit for AS patients received etanercept [63]. When the inhibition of progression of structural damage in the spine was measured by the mean change in the modified Stoke AS Spine Score (mSASSS) (range 0–72) from baseline to 2 years, the failure of inhibition of structural damage progression in AS was confirmed also for adalimumab [69] and infliximab [70].
A number of recent studies have evaluated peripheral joint destruction in AS patients on TNF blocking agents and found that these agents are effective in reducing structural deterioration. These data are summarized in a recent publication [71].
Interleukin-17 Inhibition with AS
Recently, the Th17 T cell and the cytokine it produces, interleukin-17 (IL-17), have been reported to increase joint inflammation and induce osteoclast differentiation and bone destruction in autoimmune arthritis [72]. Preclinical studies have reported that IL-17 can stimulate synoviocytes to produce RANKL and activate osteoclastogenesis [73]. Secukinumab is a fully human IgG1 monoclonal antibody that selectively binds to and neutralizes IL-17A. Secukinumab is approved for the treatment of moderate-to-severe AS. Two phase III studies evaluated the efficacy and safety of secukinumab (loading dose 10 mg/kg baseline, weeks 2 and 4) followed by either 300 mg every 4 weeks or 150 mg every week for 4 weeks. After 16 weeks, significant improvements in the AS International Society criteria for 20% improvement (ASAS20) were nearly 60% for both secukinumab IV-300 mg and IV-150 mg compared to placebo at 37%, and the improvements were maintained at week 52 [74]. Other studies of secukinumab in AS have reported similar results [75, 76]. Currently, there are no studies with secukinumab that evaluated localized or systemic bone mass changes; however, the reduction in structural changes within the inflamed joints suggests that bone mass may be improved with the treatment.
Osteoporosis and Psoriatic Arthritis (PsA)
PsA is a systemic inflammatory arthritis in which subjects with a long disease duration can have both localized and systemic bone loss secondary to systemic inflammation, medications used to treat the disease and reduced activity related to joint pain. A cross-sectional study of BMD in patients with PsA reported that low bone mass and osteoporosis were associated with longer average duration of disease [77]. Another cross-sectional and longitudinal study of PsA subjects found the frequency of low BMD and osteoporosis no different from the general population [78].
The Skeletal Phenotype of PsA
The skeletal phenotype of PsA patients is more similar to AS than RA. PsA is associated with increased bone formation at the sites of inflammation. The bone formation in the axial skeleton can take the form of marginal or paramarginal syndesmophytes that are asymmetrically distributed, and sacroiliitis that is frequently asymmetric. In the peripheral skeleton bone formation can be present in the periosteum within the joints as ankylosis, and within the enthesis [7].
The biology of the bone changes in PsA is an active area of research and is reviewed in detail by Paine and Richlin [7]. Briefly, in PsA, there is altered bone remodeling due to an increase in inflammatory cytokines including TNF, IL-1, and Il-17, growth factors, and ill-defined biomechanical factors. Inflammatory cytokines including TNF alpha and IL-17 are released from the synovitis, and stimulate osteoclast maturation and activity indirectly through RANKL production or directly by stimulating osteoclast activity. However, simultaneously, the localized production of IL-23 in the inflamed enthesis can stimulate IL-22 and BMP production, thereby stimulating new bone formation in the inflamed joint [7, 66]. The biology of the bone changes in PsA have informed clinicians about optimal treatments.
Conventional DMARDs and Bone in PsA
There are no studies of the effects of PsA on localized or systemic bone mass in PsA.
Biologic Agents, Bone Mass, and PsA
There are no studies that have evaluated the effect of biologic agents used to treat the inflammatory joint activity in patients with PsA on localized or systemic bone mass. However, there are now seven biologic agents and apremilast FDA approved to inhibit structural damage from PsA, and these studies have been reviewed recently [7]. While all agents, but apremilast, have demonstrated an ability to slow radiographic progression, none have been able to inhibit the new bone formation [7]. Interestingly, many years ago, it was shown that treatment with non-steroidal anti-inflammatory agents (NSAIDs) was able to inhibit the new bone formation in AS patients [79], and additional studies are needed to determine if the NSAIDs may be effective in PsA.
IL-12/23 and Interleukin-17 Inhibition with PsA
While a number of new biologic agents are approved for the treatment of PsA, nearly all of these agents were originally studied and received approval in RA. However, the inhibition of IL-17 and IL-12/23 and IL-23 were initially studied in PsA and we will review the studies briefly.
Interleukin-23 (IL-23) is an inflammatory cytokine and expression is increased in animal models of arthritis. It is associated with enthesitis via activation of T cells that produce Il-17, TNF, and Il-22. A monoclonal antibody to IL-12/23, ustekinumab, was studied in patients with active PsA, and it slowed radiographic progression both in TNF previously treated or TNF naïve PsA subjects [80,81,82].
Secukinumab is a fully human IgG1 monoclonal antibody that selectively binds to and neutralizes IL-17A. Secukinumab is approved for the treatment of moderate-to-severe psoriasis, PsA. Secukinumab significantly reduced radiographic progression, measured by change from baseline to week 24 in the modified total Sharp/van der Heijde score (SHS) for PsA compared with placebo [83]. In a phase III study of active PsA, radiographic progression using SHS for PsA was inhibited in all secukinumab-treated patients through 52 weeks [84]. Currently, there are no studies with secukinumab that evaluated localized or systemic bone mass changes; however, the reduction in structural changes within the inflamed joints suggests that bone mass may be improved with the treatment.
Anti-osteoporotic Medication in Rheumatic Disease Patients
Bisphosphonates
Bisphosphonates are widely used as anti-osteoporotic medication for decades. Recently, its role for a structural benefit in RA has been highlighted. A study on early RA randomized 39 patients to receive zoledronic acid or placebo, with both groups receiving MTX. Patients were evaluated at 26 weeks for MRI measured hand and wrist erosion, and were 61% lower in the zoledronic acid + MTX group compared to the placebo + MTX group [85]. Also, PsA patients treated with zoledronic acid had a significant reduced MRI measured bone edema [86]. Further studies on bisphosphonates and DMARDs are warranted.
Denosumab
RANK/RANKL/OPG pathway plays an important role in osteoclast differentiation, activity, and survival. OPG is a natural inhibitor of RANKL, and the idea of inhibition of RANKL led to the development of denosumab, a fully human antibody that binds to, and inhibits, RANKL [87]. In a study of RA patients with active disease treated with MTX, denosumab successfully reduced radiographic progression and improved BMD of lumbar spine and total hip 6 and 12 months after treatment, regardless of concomitant glucocorticoid administration, while bone turnover markers were suppressed [88]. In the denosumab treatment group receiving 180 mg of denosumab and no glucocorticoids, BMD at the lumbar spine increased by 2% compared to placebo at 6 months, and 4% at 12 months. In the same treatment group also receiving glucocorticoids, lumbar spine BMD increased by approximately 2.4% at 6 months, and 3.2% at 12 months, compared to placebo. Measurements of total hip BMD showed 1 and 1.7% at 6 and 12 months, respectively, for denosumab without glucocorticoids, and 1.2 and 1.7% for denosumab with glucocorticoids [88].
In a randomized clinical trial, 40 patients with RA received either alendronate or denosumab for 6 months. A post-hoc analysis of changes in bony erosions at the second metatarsal head reported a significant reduction in bone erosion in the denosumab group, but a significant increase in erosions in the alendronate-treated group [89]. However, denosumab had negligible effects on the RA disease activity based on the American College of Rheumatology (ACR) response parameters [90, 91].
Treatment with denosumab was also shown to decrease joint erosion measured with MRI. Metacarpophalangeal joints and wrists were analyzed on 218 patients receiving either placebo or denosumab for 12 months. Patients receiving 180 mg of denosumab showed a decrease in joint erosion measured by MRI at 6 months after treatment; however, those treated with 60 mg showed no significant improvement [91]. 12 months after treatment, patients treated with denosumab showed a significant decrease in the total modified Sharp erosion scores, but no difference in the modified Sharp joint space narrowing score. In addition, bone turnovers markers, serum CTX-1 and serum PINP, significantly decreased in denosumab treated groups compared to the placebo groups at 12 months [91]. An additional analysis of the same study measured lumbar spine and hip BMD and denosumab significantly increased the BMD at the lumbar spine and hip compared to the placebo group at 12 months [91].
Recent publications report that cessation of denosumab therapy can lead to a rapid decrease of BMD, thus increasing risk of fracture [92]. Therefore, it is recommended that either denosumab should be continued without drug holiday, or denosumab should be switched to other osteoporotic medication upon discontinuation, even if BMD is well above values recommended for treatment.
Parathyroid Hormone (PTH) and Analogs
PTH exerts its efficacy depending the route of administration. The continuous administration of PTH leads to increased bone remodeling with bone loss, whereas intermittent, daily, low-dose administration of PTH has an osteoanabolic effect [93]. Since PTH can stimulate bone formation and glucocorticoids reduce bone formation, rheumatic disease patients on chronic glucocorticoids have been treated with PTH. Lane et al. studied osteoporotic postmenopausal women with rheumatic diseases on glucocorticoids and determined that hPTH (1–34) and estrogen increased bone mass at the lumbar spine and hip after 12 months of treatment significantly more than estrogen alone [94]. Saag et al. compared alendronate to teriparatide in glucocorticoid-induced osteoporosis and reported teriparatide was associated with increased bone mineral density at the lumbar spine and total hip with reduced incidence of new vertebral fracture compared with alendronate [95]. Another study in glucocorticoid treated men found treatment with hPTH (1–34) had greater improvements in BMD, microstructure and bone strength than risedronate [96]. Moreover, patients with RA showed greater response to daily hPTH (1–34) administration at the femoral neck at 18 months than postmenopausal osteoporotic patients, and there was a positive effect of biologics combined with teriparatide in elevating bone formation markers [97]. However, a pilot study randomized 20 patients to receive etanercept or etanercept + teriparatide and evaluated patients at 12 months for changes in arthritis erosions. They reported that concomitant administration of etanercept and PTH did not affect healing erosions in early RA patients [98]. Also, another study evaluated the effect of teriparatide on joint erosions in RA patients and reported no significant reduction in erosion volume of hand or wrists following 1 year of treatment with teriparatide or placebo [99].
Conclusion
Inflammatory arthritis results in both generalized and localized changes in bone mass and bone structure. While biologic agents appear to reduce structural deterioration, and periarticular bone loss, the effects on systemic bone mass are limited. Blockade of TNF appears to maintain or modestly increase localized and systemic BMD in subjects with inflammatory arthritis. However, other non-TNF biologic agents approved for rheumatic diseases effects on bone have not been well studied. Additional studies that include biochemical markers of bone turnover, bone density, and fracture outcomes are needed to better inform the medical community of the long-term impact of these medications on bone health in subjects with rheumatic diseases.
References
Dequeker J, Maenaut K, Verwilghen J, Westhovens R (1995) Osteoporosis in rheumatoid arthritis. Clin Exp Rheumatol 13(Suppl 12):S21–S26
Will R, Palmer R, Bhalla AK, Ring F, Calin A (1989) Osteoporosis in early ankylosing spondylitis: a primary pathological event? Lancet 2:1483–1485
Ralston SH, Urquhart GD, Brzeski M, Sturrock RD (1990) Prevalence of vertebral compression fractures due to osteoporosis in ankylosing spondylitis. BMJ 300:563–565
Kim SY, Schneeweiss S, Liu J, Daniel GW, Chang CL, Garneau K, Solomon DH (2010) Risk of osteoporotic fracture in a large population-based cohort of patients with rheumatoid arthritis. Arthritis Res Ther 12:R154
Xue AL, Wu SY, Jiang L, Feng AM, Guo HF, Zhao P (2017) Bone fracture risk in patients with rheumatoid arthritis: a meta-analysis. Medicine 96:e6983
Davey-Ranasinghe N, Deodhar A (2013) Osteoporosis and vertebral fractures in ankylosing spondylitis. Curr Opin Rheumatol 25:509–516
Paine A, Ritchlin C (2018) Altered bone remodeling in psoriatic disease: new insights and future directions. Calcif Tissue Int 1–16
Vis M, Guler-Yuksel M, Lems WF (2013) Can bone loss in rheumatoid arthritis be prevented? Osteoporos Int 24:2541–2553
Geusens P, Lems WF (2011) Osteoimmunology and osteoporosis. Arthritis Res Ther 13:242
Geusens P (2012) The role of RANK ligand/osteoprotegerin in rheumatoid arthritis. Ther Adv Musculoskelet Dis 4:225–233
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, Korb A, Smolen J, Hoffmann M, Scheinecker C, van der Heide D, Landewe R, Lacey D, Richards WG, Schett G (2007) Dickkopf-1 is a master regulator of joint remodeling. Nat Med 13:156–163
Haugeberg G, Uhlig T, Falch JA, Halse JI, Kvien TK (2000) Bone mineral density and frequency of osteoporosis in female patients with rheumatoid arthritis: results from 394 patients in the Oslo County Rheumatoid Arthritis register. Arthritis Rheum 43:522–530
Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E (2008) FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 19:385–397
Confavreux CB, Chapurlat RD (2011) Systemic bone effects of biologic therapies in rheumatoid arthritis and ankylosing spondylitis. Osteoporos Int 22:1023–1036
Engvall IL, Svensson B, Boonen A, van der Heijde D, Lerner UH, Hafstrom I (2013) Low-dose prednisolone in early rheumatoid arthritis inhibits collagen type I degradation by matrix metalloproteinases as assessed by serum 1CTP: a possible mechanism for specific inhibition of radiological destruction. Rheumatology 52:733–742
Yao W, Cheng Z, Busse C, Pham A, Nakamura MC, Lane NE (2008) Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: a longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthritis Rheum 58:1674–1686
Zerbini CAF, Clark P, Mendez-Sanchez L, Pereira RMR, Messina OD, Una CR, Adachi JD, Lems WF, Cooper C, Lane NE (2017) Biologic therapies and bone loss in rheumatoid arthritis. Osteoporos Int 28:429–446
Goldring SR (2016) Differential mechanisms of de-regulated bone formation in rheumatoid arthritis and spondyloarthritis. Rheumatology 55:ii56-ii60
Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G (2005) Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 8:751–764
Both T, Zillikens MC, Schreuders-Koedam M, Vis M, Lam WK, Weel A, van Leeuwen J, van Hagen PM, van der Eerden BCJ, van Daele PLA (2018) Hydroxychloroquine affects bone resorption both in vitro and in vivo. J Cell Physiol 233:1424–1433
Tengstrand B, Hafstrom I (2002) Bone mineral density in men with rheumatoid arthritis is associated with erosive disease and sulfasalazine treatment but not with sex hormones. J Rheumatol 29:2299–2305
Pfeil A, Lippold J, Eidner T, Lehmann G, Oelzner P, Renz DM, Hansch A, Wolf G, Hein G, Kaiser WA, Bottcher J (2009) Effects of leflunomide and methotrexate in rheumatoid arthritis detected by digital X-ray radiogrammetry and computer-aided joint space analysis. Rheumatol Int 29:287–295
Kaste SC, Jones-Wallace D, Rose SR, Boyett JM, Lustig RH, Rivera GK, Pui CH, Hudson MM (2001) Bone mineral decrements in survivors of childhood acute lymphoblastic leukemia: frequency of occurrence and risk factors for their development. Leukemia 15:728–734
di Munno O, Mazzantini M, Sinigaglia L, Bianchi G, Minisola G, Muratore M, la Corte R, di Matteo L, Canesi B, Caminiti M, Broggini M, Adami S (2004) Effect of low dose methotrexate on bone density in women with rheumatoid arthritis: results from a multicenter cross-sectional study. J Rheumatol 31:1305–1309
Cranney AB, McKendry RJ, Wells GA, Ooi DS, Kanigsberg ND, Kraag GR, Smith CD (2001) The effect of low dose methotrexate on bone density. J Rheumatol 28:2395–2399
Minaur NJ, Kounali D, Vedi S, Compston JE, Beresford JN, Bhalla AK (2002) Methotrexate in the treatment of rheumatoid arthritis. II. In vivo effects on bone mineral density. Rheumatology 41:741–749
Tascioglu F, Oner C, Armagan O (2003) The effect of low-dose methotrexate on bone mineral density in patients with early rheumatoid arthritis. Rheumatol Int 23:231–235
Carbone LD, Kaeley G, McKown KM, Cremer M, Palmieri G, Kaplan S (1999) Effects of long-term administration of methotrexate on bone mineral density in rheumatoid arthritis. Calcif Tissue Int 64:100–101
Bianchi ML, Cimaz R, Galbiati E, Corona F, Cherubini R, Bardare M (1999) Bone mass change during methotrexate treatment in patients with juvenile rheumatoid arthritis. Osteoporos Int 10:20–25
Buckley LM, Leib ES, Cartularo KS, Vacek PM, Cooper SM (1997) Effects of low dose methotrexate on the bone mineral density of patients with rheumatoid arthritis. J Rheumatol 24:1489–1494
Torikai E, Kageyama Y, Takahashi M, Nagano A (2006) The effect of methotrexate on bone metabolism markers in patients with rheumatoid arthritis. Mod Rheumatol 16:350–354
Rexhepi S, Rexhepi M, Sahatciu-Meka V, Mahmutaj V, Boshnjaku S (2016) The impact of low-dose disease-modifying anti-rheumatics drugs (DMARDs) on bone mineral density of premenopausal women in early rheumatoid arthritis. Med Arch 70:101–103
Arai K, Hanyu T, Sugitani H, Murai T, Fujisawa J, Nakazono K, Kondo N, Endo N (2006) Risk factors for vertebral fracture in menopausal or postmenopausal Japanese women with rheumatoid arthritis: a cross-sectional and longitudinal study. J Bone Mineral Metab 24:118–124
Saidenberg-Kermanac’h N, Corrado A, Lemeiter D, deVernejoul MC, Boissier MC, Cohen-Solal ME (2004) TNF-alpha antibodies and osteoprotegerin decrease systemic bone loss associated with inflammation through distinct mechanisms in collagen-induced arthritis. Bone 35:1200–1207
Korczowska I, Lacki JK, Hrycaj P (2013) Influence of infliximab on cytokines network and markers of bone remodeling in rheumatoid arthritis patients. Yonsei Med J 54:183–188
Chopin F, Garnero P, le Henanff A, Debiais F, Daragon A, Roux C, Sany J, Wendling D, Zarnitsky C, Ravaud P, Thomas T (2008) Long-term effects of infliximab on bone and cartilage turnover markers in patients with rheumatoid arthritis. Ann Rheum Dis 67:353–357
Vis M, Wolbink GJ, Lodder MC, Kostense PJ, van de Stadt RJ, de Koning MH, Dijkmans BA, Lems WF (2003) Early changes in bone metabolism in rheumatoid arthritis patients treated with infliximab. Arthritis Rheum 48:2996–2997
Marotte H, Pallot-Prades B, Grange L, Gaudin P, Alexandre C, Miossec P (2007) A 1-year case-control study in patients with rheumatoid arthritis indicates prevention of loss of bone mineral density in both responders and nonresponders to infliximab. Arthritis Res Ther 9:R61
Vis M, Havaardsholm EA, Haugeberg G, Uhlig T, Voskuyl AE, van de Stadt RJ, Dijkmans BA, Woolf AD, Kvien TK, Lems WF (2006) Evaluation of bone mineral density, bone metabolism, osteoprotegerin and receptor activator of the NFkappaB ligand serum levels during treatment with infliximab in patients with rheumatoid arthritis. Ann Rheum Dis 65:1495–1499
Lange U, Teichmann J, Muller-Ladner U, Strunk J (2005) Increase in bone mineral density of patients with rheumatoid arthritis treated with anti-TNF-alpha antibody: a prospective open-label pilot study. Rheumatology 44:1546–1548
Wijbrandts CA, Klaasen R, Dijkgraaf MG, Gerlag DM, van Eck-Smit BL, Tak PP (2009) Bone mineral density in rheumatoid arthritis patients 1 year after adalimumab therapy: arrest of bone loss. Ann Rheum Dis 68:373–376
Vis M, Voskuyl AE, Wolbink GJ, Dijkmans BA, Lems WF (2005) Bone mineral density in patients with rheumatoid arthritis treated with infliximab. Ann Rheum Dis 64:336–337
Guler-Yuksel M, Bijsterbosch J, Goekoop-Ruiterman YP, de Vries-Bouwstra JK, Hulsmans HM, de Beus WM, Han KH, Breedveld FC, Dijkmans BA, Allaart CF, Lems WF (2008) Changes in bone mineral density in patients with recent onset, active rheumatoid arthritis. Ann Rheum Dis 67:823–828
Eekman DA, Vis M, Bultink IE, Kuik DJ, Voskuyl AE, Dijkmans BA, Lems WF (2011) Stable bone mineral density in lumbar spine and hip in contrast to bone loss in the hands during long-term treatment with infliximab in patients with rheumatoid arthritis. Ann Rheum Dis 70:389–390
Krieckaert CL, Nurmohamed MT, Wolbink G, Lems WF (2013) Changes in bone mineral density during long-term treatment with adalimumab in patients with rheumatoid arthritis: a cohort study. Rheumatology 52:547–553
Garnero P, Thompson E, Woodworth T, Smolen JS (2010) Rapid and sustained improvement in bone and cartilage turnover markers with the anti-interleukin-6 receptor inhibitor tocilizumab plus methotrexate in rheumatoid arthritis patients with an inadequate response to methotrexate: results from a substudy of the multicenter double-blind, placebo-controlled trial of tocilizumab in inadequate responders to methotrexate alone. Arthritis Rheum 62:33–43
Karsdal MA, Schett G, Emery P, Harari O, Byrjalsen I, Kenwright A, Bay-Jensen AC, Platt A (2012) IL-6 receptor inhibition positively modulates bone balance in rheumatoid arthritis patients with an inadequate response to anti-tumor necrosis factor therapy: biochemical marker analysis of bone metabolism in the tocilizumab RADIATE study (NCT00106522). Semin Arthritis Rheum 42:131–139
Briott K, Rouanet S, Schaeverbeke T, Etchepare F, Gaudin P, Perdriger A, Vray M, Steinberg G, Roux C (2015) The effect of tocilizumab on bone mineral density, serum levels of Dickkopf-1 and bone remodeling markers in patients with rheumatoid arthritis. Joint Bone Spine 82(2):109–115
Chen YM, Chen HH, Huang WN, Liao TL, Chen JP, Chao WC, Lin CT, Hung WT, Hsieh CW, Hsieh TY, Chen YH, Chen DY (2017) Tocilizumab potentially prevents bone loss in patients with anticitrullinated protein antibody-positive rheumatoid arthritis. PLoS ONE 12:e0188454
Kume K, Amano K, Yamada S, Kanazawa T, Ohta H, Hatta K, Amano K, Kuwaba N (2014) The effect of tocilizumab on bone mineral density in patients with methotrexate-resistant active rheumatoid arthritis. Rheumatology 53:900–903
Meyer DM, Jesson MI, Li X, Elrick MM, Funckes-Shippy CL, Warner JD, Gross CJ, Dowty ME, Ramaiah SK, Hirsch JL, Saabye MJ, Barks JL, Kishore N, Morris DL (2010) Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflamm 7:41
Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, Gruben D, Koncz T, Krishnaswami S, Wallenstein GV, Zang C, Zwillich SH, van Vollenhoven RF (2014) Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 370:2377–2386
van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, Cardiel MH, Cohen S, Nash P, Song YW, Tegzova D, Wyman BT, Gruben D, Benda B, Wallenstein G, Krishnaswami S, Zwillich SH, Bradley JD, Connell CA (2013) Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum 65:559–570
Vidal B, Cascao R, Finnila MAJ, Lopes IP, da Gloria VG, Saarakkala S, Zioupos P, Canhao H, Fonseca JE (2017) Effects of tofacitinib in early arthritis-induced bone loss in an adjuvant-induced arthritis rat model. Rheumatology
Chen XD, Xiao P, Lei SF, Liu YZ, Guo YF, Deng FY, Tan LJ, Zhu XZ, Chen FR, Recker RR, Deng HW (2010) Gene expression profiling in monocytes and SNP association suggest the importance of the STAT1 gene for osteoporosis in both Chinese and Caucasians. J Bone Mineral Res 25:339–355
Zhou H, Newnum AB, Martin JR, Li P, Nelson MT, Moh A, Fu XY, Yokota H, Li J (2011) Osteoblast/osteocyte-specific inactivation of Stat3 decreases load-driven bone formation and accumulates reactive oxygen species. Bone 49:404–411
Hein G, Eidner T, Oelzner P, Rose M, Wilke A, Wolf G, Franke S (2011) Influence of Rituximab on markers of bone remodeling in patients with rheumatoid arthritis: a prospective open-label pilot study. Rheumatol Int 31:269–272
Boumans MJ, Thurlings RM, Yeo L, Scheel-Toellner D, Vos K, Gerlag DM, Tak PP (2012) Rituximab abrogates joint destruction in rheumatoid arthritis by inhibiting osteoclastogenesis. Ann Rheum Dis 71:108–113
Salvin SQI, Master M, Corazza I, De Marchi G, Lombardi S et al (2010) Variations in lumbar spine and femoral BMD after rituximab therapy in active rheumatoid arthritis. Ann Rheum Dis 69:704
Axmann R, Herman S, Zaiss M, Franz S, Polzer K, Zwerina J, Herrmann M, Smolen J, Schett G (2008) CTLA-4 directly inhibits osteoclast formation. Ann Rheum Dis 67:1603–1609
Tada M, Inui K, Sugioka Y, Mamoto K, Okano T, Koike T, Nakamura H (2015) FRI0062 influence of biologic agents on bone mineral density and bone mineral markers in patients with rheumatoid arthritis: data from the airtight study. Ann Rheum Dis 74:441–442
Pray C, Feroz NI, Nigil Haroon N (2017) Bone mineral density and fracture risk in ankylosing spondylitis: a meta-analysis. Calcif Tissue Int 101:182–192
van der Heijde D, Landewe R, Einstein S, Ory P, Vosse D, Ni L, Lin SL, Tsuji W, Davis JC Jr (2008) Radiographic progression of ankylosing spondylitis after up to two years of treatment with etanercept. Arthritis Rheum 58:1324–1331
Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, Gorman DM, Bowman EP, McClanahan TK, Yearley JH, Eberl G, Buckley CD, Kastelein RA, Pierce RH, Laface DM, Cua DJ (2012) IL-23 induces spondyloarthropathy by acting on ROR-gammat + CD3 + CD4-CD8- entheseal resident T cells. Nat Med 18:1069–1076
El-Zayadi AA, Jones EA, Churchman SM, Baboolal TG, Cuthbert RJ, El-Jawhari JJ, Badawy AM, Alase AA, El-Sherbiny YM, McGonagle D (2017) Interleukin-22 drives the proliferation, migration and osteogenic differentiation of mesenchymal stem cells: a novel cytokine that could contribute to new bone formation in spondyloarthropathies. Rheumatology 56:488–493
Lories RJ, Haroon N (2014) Bone formation in axial spondyloarthritis. Best Pract Res Clin Rheumatol 28:765–777
Brandt J, Haibel H, Cornely D, Golder W, Gonzalez J, Reddig J, Thriene W, Sieper J, Braun J (2000) Successful treatment of active ankylosing spondylitis with the anti-tumor necrosis factor alpha monoclonal antibody infliximab. Arthritis Rheum 43:1346–1352
Allali F, Breban M, Porcher R, Maillefert JF, Dougados M, Roux C (2003) Increase in bone mineral density of patients with spondyloarthropathy treated with anti-tumour necrosis factor alpha. Ann Rheum Dis 62:347–349
Visvanathan S, van der Heijde D, Deodhar A, Wagner C, Baker DG, Han J, Braun J (2009) Effects of infliximab on markers of inflammation and bone turnover and associations with bone mineral density in patients with ankylosing spondylitis. Ann Rheum Dis 68:175–182
Kang KY, Lee KY, Kwok SK, Ju JH, Park KS, Hong YS, Kim HY, Park SH (2011) The change of bone mineral density according to treatment agents in patients with ankylosing spondylitis. Joint Bone Spine 78:188–193
Szentpetery A, Horvath A, Gulyas K, Petho Z, Bhattoa HP, Szanto S, Szucs G, FitzGerald O, Schett G, Szekanecz Z (2017) Effects of targeted therapies on the bone in arthritides. Autoimmun Rev 16:313–320
Hashimoto M (2017) Th17 in animal models of rheumatoid arthritis. J Clin Med 6:73
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H (2006) Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 203:2673–2682
Pavelka K, Kivitz A, Dokoupilova E, Blanco R, Maradiaga M, Tahir H, Pricop L, Andersson M, Readie A, Porter B (2017) Efficacy, safety, and tolerability of secukinumab in patients with active ankylosing spondylitis: a randomized, double-blind phase 3 study, MEASURE 3. Arthritis Res Ther 19:285
Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, van der Heijde D, McInnes I, van Laar JM, Landewe R, Wordsworth P, Wollenhaupt J, Kellner H, Paramarta J, Wei J, Brachat A, Bek S, Laurent D, Li Y, Wang YA, Bertolino AP, Gsteiger S, Wright AM, Hueber W (2013) Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet 382:1705–1713
Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, Deodhar A, Porter B, Martin R, Andersson M, Mpofu S, Richards HB (2015) Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med 373:2534–2548
D’Epiro S, Marocco C, Salvi M, Mattozzi C, Luci C, Macaluso L, Giancristoforo S, Campoli M, Scarno M, Migliaccio S, Calvieri S, Richetta A (2014) Psoriasis and bone mineral density: implications for long-term patients. J Dermatol 41:783–787
Busquets N, Vaquero CG, Moreno JR, Vilaseca DR, Narvaez J, Carmona L, Nolla JM (2014) Bone mineral density status and frequency of osteoporosis and clinical fractures in 155 patients with psoriatic arthritis followed in a university hospital. Reumatol Clin 10:89–93
Wanders A, Heijde D, Landewe R, Behier JM, Calin A, Olivieri I, Zeidler H, Dougados M (2005) Nonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis: a randomized clinical trial. Arthritis Rheum 52:1756–1765
McInnes IB, Kavanaugh A, Gottlieb AB, Puig L, Rahman P, Ritchlin C, Brodmerkel C, Li S, Wang Y, Mendelsohn AM, Doyle MK (2013) Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 382:780–789
Kavanaugh A, Mease PJ, Gomez-Reino JJ, Adebajo AO, Wollenhaupt J, Gladman DD, Lespessailles E, Hall S, Hochfeld M, Hu C, Hough D, Stevens RM, Schett G (2014) Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis 73:1020–1026
Ritchlin C, Rahman P, Kavanaugh A, McInnes IB, Puig L, Li S, Wang Y, Shen YK, Doyle MK, Mendelsohn AM, Gottlieb AB (2014) Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann Rheum Dis 73:990–999
Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, Landewe R, Nash P, Pricop L, Yuan J, Richards HB, Mpofu S (2015) Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N Engl J Med 373:1329–1339
van der Heijde D, Landewe RB, Mease PJ, McInnes IB, Conaghan PG, Pricop L, Ligozio G, Richards HB, Mpofu S (2016) Brief Report: secukinumab provides significant and sustained inhibition of joint structural damage in a phase III study of active psoriatic arthritis. Arthritis Rheumatol 68:1914–1921
Jarrett SJ, Conaghan PG, Sloan VS, Papanastasiou P, Ortmann CE, O’Connor PJ, Grainger AJ, Emery P (2006) Preliminary evidence for a structural benefit of the new bisphosphonate zoledronic acid in early rheumatoid arthritis. Arthritis Rheum 54:1410–1414
McQueen F, Lloyd R, Doyle A, Robinson E, Lobo M, Exeter M, Taylor WJ, Jones P, Reid IR, Dalbeth N (2011) Zoledronic acid does not reduce MRI erosive progression in PsA but may suppress bone oedema: the zoledronic acid in psoriatic arthritis (ZAPA) Study. Ann Rheum Dis 70:1091–1094
Bandeira L, Bilezikian JP (2017) Novel therapies for postmenopausal osteoporosis. Endocrinol Metab Clin North Am 46:207–219
Dore RK, Cohen SB, Lane NE, Palmer W, Shergy W, Zhou L, Wang H, Tsuji W, Newmark R (2010) Effects of denosumab on bone mineral density and bone turnover in patients with rheumatoid arthritis receiving concurrent glucocorticoids or bisphosphonates. Ann Rheum Dis 69:872–875
Yue J, Griffith JF, Xiao F, Shi L, Wang D, Shen J, Wong P, Li EK, Li M, Li TK, Zhu TY, Hung VW, Qin L, Tam LS (2017) Repair of bone erosion in rheumatoid arthritis by denosumab: a high-resolution peripheral quantitative computed tomography study. Arthritis Care Res 69:1156–1163
Takeuchi T, Tanaka Y, Ishiguro N, Yamanaka H, Yoneda T, Ohira T, Okubo N, Genant HK, van der Heijde D (2016) Effect of denosumab on Japanese patients with rheumatoid arthritis: a dose-response study of AMG 162 (Denosumab) in patients with RheumatoId arthritis on methotrexate to Validate inhibitory effect on bone Erosion (DRIVE)-a 12-month, multicentre, randomised, double-blind, placebo-controlled, phase II clinical trial. Ann Rheum Dis 75:983–990
Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, van der Heijde D, Zhou L, Tsuji W, Newmark R (2008) Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum 58:1299–1309
Cummings SR, Ferrari S, Eastell R, Gilchrist N, Jensen JB, McClung M, Roux C, Torring O, Valter I, Wang AT, Brown JP (2017) Vertebral fractures after discontinuation of denosumab: a post hoc analysis of the randomized placebo-controlled FREEDOM trial and its extension. J Bone Mineral Res
Canalis E, Giustina A, Bilezikian JP (2007) Mechanisms of anabolic therapies for osteoporosis. N Engl J Med 357:905–916
Lane NE, Sanchez S, Modin GW, Genant HK, Pierini E, Arnaud CD (1998) Parathyroid hormone treatment can reverse corticosteroid-induced osteoporosis. Results of a randomized controlled clinical trial. J Clin Invest 102:1627–1633
Saag KG, Shane E, Boonen S, Marin F, Donley DW, Taylor KA, Dalsky GP, Marcus R (2007) Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 357:2028–2039
Gluer CC, Marin F, Ringe JD, Hawkins F, Moricke R, Papaioannu N, Farahmand P, Minisola S, Martinez G, Nolla JM, Niedhart C, Guanabens N, Nuti R, Martin-Mola E, Thomasius F, Kapetanos G, Pena J, Graeff C, Petto H, Sanz B, Reisinger A, Zysset PK (2013) Comparative effects of teriparatide and risedronate in glucocorticoid-induced osteoporosis in men: 18-month results of the EuroGIOPs trial. J Bone Mineral Res 28:1355–1368
Ebina K, Hashimoto J, Shi K, Kashii M, Hirao M, Yoshikawa H (2014) Comparison of the effect of 18-month daily teriparatide administration on patients with rheumatoid arthritis and postmenopausal osteoporosis patients. Osteoporos Int 25:2755–2765
Migliore A, Massafra U, Bizzi E, Argento G, Diamanti AP, Germano V, Tormenta S, Arduini F, Iannessi F, Granatas M, Lagana B (2012) May etanercept and PTH (1–34) association heal erosions in early rheumatoid arthritis? A pilot study. Eur Rev Med Pharmacol Sci 16:363–369
Solomon DH, Kay J, Duryea J, Lu B, Bolster MB, Yood RA, Han R, Ball S, Coleman C, Lo E, Wohlfahrt A, Sury M, Yin M, Yu Z, Zak A, Gravallese EM (2017) Effects of teriparatide on joint erosions in rheumatoid arthritis: a randomized controlled trial. Arthritis Rheumatol 69:1741–1750
Acknowledgements
This work was supported in part by Inha University, Incheon, Korea, endowment for Dr. Nancy E. Lane at UC Davis, NIH Grant P50 AR063043 (NEL).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Alanna M. Dubrovsky, Mie Jin Lim, Nancy E. Lane have no conflicts of interest related to this work.
Human and Animal Rights and Informed Consent
There were no human or animal studies that were performed by the authors that were described in this work.
Rights and permissions
About this article

Cite this article
Dubrovsky, A.M., Lim, M.J. & Lane, N.E. Osteoporosis in Rheumatic Diseases: Anti-rheumatic Drugs and the Skeleton. Calcif Tissue Int 102, 607–618 (2018). https://doi.org/10.1007/s00223-018-0401-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-018-0401-9