
Abstract
Sarcopenia and osteoporosis are two sides of the same coin. They represent different aspects of the same age-related process of musculoskeletal atrophy and together culminate in falls, fractures, deconditioning, and increased mortality in older individuals. However, the current therapeutic approach to the prevention of minimal trauma fracture is unilateral and focuses solely on bone. In theory, an integrated approach that recognizes the interaction between muscle and bone could break the vicious cycle of their combined involution and more effectively minimize falls/fractures. In this review, signaling pathways and cross-talk mechanisms that integrate bone/muscle, and the emergence of novel therapies that exploit these pathways to target osteoporosis/sarcopenia will be discussed. In broad terms, these agents act on nuclear receptors (e.g., VDR, AR) or transmembrane receptors (e.g., activins, GH/IGF-1) expressed in muscle and bone, and seek to alter biologic responses to musculoskeletal aging, loading, and injury. Challenges in the development of these dual bone–muscle therapies, early clinical trials examining their safety/efficacy, and novel targets that hold promise in the reversal of musculoskeletal aging will be discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Musculoskeletal disorders are very common and affect up to 50 % of individuals in western countries [1, 2]. These diseases are also costly and lead to an annual expenditure of approximately $850 billion in the United States (8 % GDP) and $4.5 billion in Australia (10 % GDP). As the world’s population ages, the concerning effects of musculoskeletal wasting will be on the rise: falls, fractures, the need for assisted care, and the financial impact of these significant life events [3].
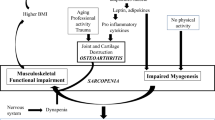
Reduced muscle mass affects balance, increases postural sway and thereby increases the risk of falls. As the subject falls, fractures result due to bone’s inability to withstand the loading forces applied to it by muscle and the subsequent trauma [4]. In this regard, the gradual age-related decline in bone and muscle (i.e., osteoporosis and sarcopenia) culminate in acute events with potentially catastrophic effects on morbidity and mortality [5].
For many years, therapies used to prevent minimal trauma fractures have focused solely on bone. Muscle and, more specifically, the effects of sarcopenia in compounding the risk of osteoporotic fracture have not received due attention. As we begin to recognize the tight link between sarcopenia and osteoporosis [6] and the effects of muscle mass on fracture risk [7], a paradigm shift in the treatment of these conditions may be underway. Recently, experts have suggested a more inclusive name be given to the combination of sarcopenia and osteoporosis, such as the “dysmobility syndrome”, which integrates their pathogenesis and unites them as a single therapeutic target [8].
There are, however, technical challenges in considering osteoporosis and sarcopenia as a single entity. Osteoporosis has been clearly defined but sarcopenia and its endpoints remain elusive [9]. Although there is consensus that a combination of functional and quantitative parameters of muscle mass are necessary to define sarcopenia [10–12], the correlation of specific muscle tests to clinically meaningful outcomes remains an open question [13].
Another challenge is to better understand the interconnected biology of the musculoskeletal system. We have known for centuries that muscle and bone interact anatomically and through complex biomechanical signals. More recently, a sophisticated network of paracrine and endocrine signals that coordinates bone and muscle has emerged with integrated effects in development, the response to injury and aging [14]. There has been a call for greater research in bone–muscle interactions and the development of therapeutic agents that target sarcopenia and osteoporosis as a single disorder [5]. A holistic approach to these conditions may be a more effective one, directly addressing the mechanisms of osteoporotic fracture via bone and its fundamentally important neighbor, muscle [15]. In this review, the biological basis of bone and muscle interactions, therapies that target muscle and bone as a single unit, and future directions in this exciting field of research will be discussed.
Bone and Muscle Interactions
Throughout life’s various stages, the musculoskeletal system operates as a finely coordinated unit. Muscle and bone originate from the mesenchymal progenitor cell during embryonic life and their development is regulated by overlapping genes and humoral factors [16, 17]. Mechanical forces from developing muscles exert critical effects in periosteal bone growth, bone geometry, and density. In demonstrating this integral association between bone and muscle development, children with Duchenne muscular dystrophy (DMD) and cerebral palsy have reduced bone density and a greater fracture risk [18, 19]. Mice with congenital paralysis have severe impairments in bone development and mineralization [20, 21].
Throughout post-natal life, sex steroids, insulin-like growth factor-1 (IGF-1), and growth hormone (GH) coordinate musculoskeletal growth. Muscle seems to possess the “upper hand” in its relationship with bone, driving changes in bone density. This is seen initially in puberty where the accumulation of lean mass precedes gains in bone mass, and muscle mass determines cortical bone area [22, 23]. Conversely, in aging adults, lean mass declines before bone mass, and muscle mass correlates tightly with reductions in bone density [24, 25]. A potential explanation for muscle’s apparent primacy in these interactions is found in the “mechanostat theory”. This theory, as recently reviewed in this journal [26], proposes that muscle loading induces a range of biomechanical signals necessary for bone growth and remodeling. Indeed, subjects exposed to a gravity-free environment, such as astronauts or mice suspended in mid-air for prolonged periods, experience dramatic bone loss due to lack of muscle loading [27, 28]. Pathways responsible for mechanotransduction such as stretch-activated cation channels, G protein-coupled receptors, sclerostin, and LRP5 provide a strong molecular basis for this theory and are being targeted for their therapeutic potential [29, 30].
The “mechanostat theory”, however, does not fully explain the complex nature of bone–muscle interactions. For example, the correlation between muscle mass and bone cortical thickness is seen not only in adjacent tissues but also in muscles and bones located remotely to one another [31]. This opens the possibility of cross-talk and the presence of discrete hormonal influences between these tissues. Indeed a number of muscle and bone-derived hormones (i.e., myokines and osteokines) are under active investigation for their integrative effects. Myostatin, fibroblast growth factor 2 (FGF2), interleukin 6 (IL6), and matrix metalloproteinase 2 (MMP2) are myokines with potential effects in bone [32–34]. Conversely, humoral factors made in bone, including FGF21, undercaboxylated osteocalcin, and sclerostin exert potential effects in skeletal muscle [35, 36]. There are also common pathways such as GH/IGF-1, sex steroids, and Wnt signaling that may centrally govern the bone–muscle unit, its adaptation to mechanical stimuli and injury [31, 37].
Further support for bone–muscle cross-talk is seen in the response to musculoskeletal injury. The use of muscle flaps in the surgical correction of open fractures improves bone healing in both rodents and humans [38, 39]. Conversely, the incidence of nonunion is significantly higher in fractures associated with muscle damage, as seen in compartment syndrome [40]. Therefore, healthy muscle is a positive factor for fracture healing and muscle may represent a sort of ‘outerperiosteum’, providing morphogens and growth factors to assist bone repair.
Thus, an intricate network of biomechanical and endocrine signals connects muscle and bone with integrative effects in musculoskeletal mass and function throughout life. A number of common pathways, such as the GH/IGF1, vitamin D signaling, and androgens have been investigated for their therapeutic potential in musculoskeletal diseases. Other pathways, such as myostatin/activin signaling and bone-derived factors such as undercarboxylated osteocalcin, are also being investigated for their systemic effects. Examining these individual pathways for their specific effects and therapeutic potential poses significant experimental challenges. However, a thorough understanding of the endocrine network that connects bone and muscle is critical for the discovery of therapeutic targets that may lead to a more holistic approach to the combined syndrome of osteoporosis and sarcopenia.
Vitamin D
Although history gave vitamin D the misnomer “vitamin”, the biologically active molecule (1,25(OH)2D) is a bona fide hormone with potent effects in calcium/phosphate handling, bone homeostasis, tissue development, and immunomodulation [41]. Although its function in bone mineralization and growth plate physiology is established, our understanding of its role in muscle physiology is emerging.
Vitamin D’s capacity to integrate muscle and bone is suggested by the clinical syndrome of vitamin D deficiency/rickets. As described four centuries ago, children with rickets display the combination of “flexible, waxy” bones and “flabby, toneless” muscles [42]. Likewise, adults with vitamin D deficiency display concurrent defects in bone and muscle, characterized by osteomalacia (i.e., reduced bone mineral) and type 2 muscle fiber atrophy [43]. These conditions are also seen in subjects with mutations of the vitamin D receptor (VDR), implying local genomic effects of vitamin D in the muscle–bone unit.
However, the precise mechanism of these effects has been debated. Although vitamin D may directly alter these tissues, its musculoskeletal effects are predominantly indirect and relate to systemic calcium/phosphate homeostasis [44, 45]. To demonstrate this, subjects with rickets develop muscle wasting and bone deformity after weaning, concurrent with declining levels of calcium and phosphate (Li et al. 1997; Yoshizawa et al. 1997). This is despite the early presence of the vitamin D receptor (VDR) within embryonic mesoderm [46], the tissue from which bone and muscle arise.
In bone, the VDR is expressed mainly in osteoblasts and osteocytes, where it has been shown to directly regulate mineral formation and bone mass [47]. The presence of the VDR in muscle has been controversial for many years. However, recent studies have reported its expression in mouse and human skeletal muscle and its upregulation following vitamin D supplementation in older women [48, 49]. In studies of cultured muscle cells, vitamin D treatment leads to doubling in the size of myotubes and down-regulation of myostatin, a TGF-β which negatively regulates muscle mass [50]. The potential for vitamin D to exert anabolic muscle effects was supported by a recent pilot study. In 21 older women receiving vitamin D (4,000 IU day, 4 months), there was a 30 % increase in muscle fiber size and activation of myonuclear VDR [51]. However these changes did not translate to clinically meaningful effects in muscle strength.
Vitamin D deficiency is common in the elderly, due to nutritional deficits, reduced capacity of the skin to synthesize vitamin D, and lack of sun exposure, and has been associated with sarcopenia and osteoporosis [52, 53]. At a basic level, vitamin D deficiency mimics effects of aging with increased adipose tissue infiltration, activation of muscle proteolytic pathways, osteoclastogenesis, and increased bone turnover [54]. Elderly subjects may be more vulnerable to vitamin D deficiency due to lower expression of VDR in muscle and bone [55].
Subjects living in institutions are at particular risk of vitamin D deficiency and have been shown to benefit from vitamin D supplementation with a reduction in falls and fractures [56, 57]. However, effects of vitamin D supplementation are less clear among community dwellers. Vitamin D supplementation may increase bone mineral density in such individuals but this effect is small, limited to the femoral neck, and not associated with clear reduction in fracture risk [58, 59]. Clear effects in muscle function have been difficult to establish without standardized endpoints for strength or performance-based indices in these trials [43].
The definition of vitamin D deficiency and recommended daily intakes of vitamin D remain contentious. There is also evidence of a U-shaped curve in the musculoskeletal response to vitamin D. Individuals with high and low serum vitamin D have the greatest risk of fracture and frailty compared to those with intermediate levels [60, 61]. However, defining the intermediate range at which vitamin D effects are most beneficial has been elusive. While the Institute of Medicine (IOM) recommends 25(OH)D target levels >50 nmol/l [62], the US Endocrine society advocates a higher serum target level >75 nmol/l [63]. This uncertainty is compounded by reports of a greater incidence of kidney stones and increased falls and fractures in subjects receiving megadoses of vitamin D [58, 64]. Thus, at a certain point, the dose-limiting calcemic effects of vitamin D may outweigh its potential musculoskeletal benefits.
Therapeutic VDR agonists with low calcemic potency are being used in Japan to treat osteoporosis. One such agent, eldecalcitol, has been shown to increase bone mineral density, reduce the risk of falls/fractures, and improve lower limb strength in older individuals [65, 66]. Preclinical trials support these effects, demonstrating alterations in bone microstructure, bone turnover, and locomotive ability in rodents receiving eldecalcitol [67, 68]. These effects may result from tissue-specific activation of VDR [51] and/or direct effects on calcium flux within bone and muscle cells [67]. Although promising, the drug discovery process for VDR agonists faces potential hurdles and may hamper their widespread use. To achieve tissue selectivity, a better understanding of VDR’s molecular mechanisms, its downstream responses, particularly in muscle, and effects of polymorphisms are necessary.
In summary, musculoskeletal levels of VDR decline with age concurrent with the physiological involution of muscle and bone. Evidence suggests that musculoskeletal aging responds to vitamin D at a cellular level and in human clinical studies. Therapeutic VDR agonists provide hope in reversing the effects of musculoskeletal aging and in circumventing vitamin D’s dose-limiting calcemic effects in order to directly target muscle and bone.
Myostatin and Activin-Based Therapies
Discovered in 1997, myostatin is a member of the TGF-β superfamily and a muscle-derived hormone which is a potent negative regulator of muscle mass [69]. Mutations in the myostatin gene lead to pronounced increases in muscle mass in mammalian species [69, 70]. Conversely, increases in myostatin lead to muscle wasting in chronic diseases such as HIV [71], renal failure [72], and chronic obstructive pulmonary disease [73]. To produce these effects, myostatin binds to a transmembrane receptor (i.e., activin receptor IIB, ActRIIB), activates Smad family proteins and regulates downstream signals, such as the ubiquitin–proteasome system, that govern muscle protein turnover.
Myostatin also affects bone. Myostatin polymorphisms correlate with peak bone density [74] and myostatin knockout mice display increased bone mass and mineral [32, 75] with larger callus size following osteotomy [76]. These effects on bone are predominantly indirect, related to the increased loading forces from muscle. However, direct effects are possible due to the local expression of activin receptors in bone marrow stromal cells and osteoblasts (ActRIIA) which can weakly bind myostatin [77, 78]. In vitro studies demonstrate that directly targeting activin receptors alter bone cell differentiation [79, 80].
Activin-based therapies have the potential to target both osteoporosis and sarcopenia, given the evidence that activin signaling and myostatin regulate bone and muscle mass. Several activin-based therapies have already been developed and broadly include soluble activin receptors, recombinant follistatin (i.e., an endogenous inhibitor which bioneutralizes myostatin), derivatives of follistatin and myostatin inhibitors (i.e., propeptides and neutralizing antibodies) [78]. Rodents administered with such agents demonstrate substantial increases in muscle mass [34, 79] and reversal of muscle wasting in models of cancer cachexia [81], aging [82], and androgen deficiency [83]. Animal studies also demonstrate beneficial effects of soluble activin receptors (i.e., ActRIIB-Fc and ActRIIA) on bone mass via dual anabolic–antiresorptive effects and direct influences on osteoblast activity [79, 82, 84]. However, effects differ according to the particular agent. While soluble decoy receptors exert dual effects in muscle and bone, effects of myostatin inhibitors are specific to muscle [85, 86]. This highlights differences in tissue-specific activin signaling and may allow different therapeutic approaches depending on the predominance of muscle or bone wasting.
Human clinical trials have reported promising effects of activin-based therapies. In a phase I trial, stamulumab, a recombinant human myostatin antibody (MYO-029), was generally safe in healthy adults and patients with muscular dystrophy [87]. Although stamulumab improved single muscle fiber contraction [88], anabolic effects on muscle mass were not statistically significant. Another myostatin inhibitor (AMG-745) was shown to significantly increase lean body mass in men receiving androgen deprivation therapy for prostate cancer [89]. Activin decoy receptors have also been studied for their dual effects in muscle and bone. ACE-031 (ActRIIB decoy receptor) was shown to significantly increase lean mass (3.3 %) and thigh muscle volume (5.1 %) in a double-blind, placebo-controlled study of post-menopausal women [90]. As an indication of beneficial effects on bone, ACE-031 also significantly increased bone-specific ALP and decreased C-telopeptide, as did another soluble decoy receptor (ACE-011) [90, 91]. A significant increase in bone mineral density (3.4 %) was also reported at ~100 days in a trial of 60 post-menopausal women on ACE-031 but this data appeared only on the company website (www.acceleron.pharma.com) and has not been published or peer reviewed.
A number of adverse effects have hampered the development of activin-based therapies. Reports of nosebleeds and skin telangiectasia in two trials of ACE-031 raised concerns about unrecognized, systemic side effects of inhibiting activin signaling (NCT01099761, clinicaltrials.gov) [90]. Sudden drops in serum FSH levels after ACE-031 and ACE-011, probably related to suppression of GnRH signaling [90, 91], also raised concerns and the need for larger and longer trials to examine the safety of these therapies.
In summary, activin signaling is an attractive therapeutic target in treating osteoporosis and sarcopenia. Preclinical trials have demonstrated anabolic musculoskeletal effects of activin-based therapies and preliminary human trials confirm these effects, particularly in muscle mass. However the clinical trials are generally small, inadequately powered to detect changes in muscle strength, and too short to show convincing effects on bone density. Thus, longer and larger studies are needed to assess well-defined functional endpoints of activin-based therapies and to address concerns about their long-term safety.
Growth Hormone (GH), Insulin-Like Growth Factor 1 (IGF-1), and GH secretagogues
The GH/IGF1 axis plays a central role in musculoskeletal growth and maturation during childhood and puberty. Throughout life, it exerts system-wide effects on lipid and glucose homeostasis [92], body composition, and bone mineralization [93]. GH is a polypeptide hormone secreted in a pulsatile fashion by the anterior pituitary gland under the control of central (GHRH) and peripheral signals (IGF-1) [94]. GH acts via specific growth hormone receptors in peripheral tissues or indirectly through the induction of IGF-1 [94]. Circulating IGF-1 is synthesized mainly in the liver but IGF-1 is also expressed in numerous tissues, suggesting that local paracrine effects may be responsible for growth. At a molecular level, GH/IGF-1 signaling relies on JAK/STAT, PI3 K, and ERK pathways, and tissue-specific effects of GH versus IGF-1 illustrate the complexity of this system [95, 96]. In muscle, for example, GH determines muscle cell proliferation, fiber size, and insulin sensitivity while IGF-1 exerts effects on fiber type and is locally induced following exercise and injury [94]. In bone, GH and IGF-1 synergistically promote osteoblast activity, inhibit osteoclast-mediated bone resorption, and regulate the renal synthesis of 1,25(OH)2D and phosphate reabsorption [97–100].
GH deficiency and mutations of the GH receptor (i.e., Laron syndrome) are characterized by failure of longitudinal bone growth with short stature, failure of growth plate fusion, and reduced muscle mass, all of which respond to GH or IGF-1 replacement [101]. During aging, serum levels of GH and IGF-1 decline, correlating with reductions in muscle/bone mass and a greater risk of osteoporotic fracture [102]. In muscle, aging leads to attenuation of growth hormone receptor (GHR) activity and of exercise-mediated IGF-1 signals by the local dysregulation of miRNAs [103, 104]. Similarly, bone’s sensitivity to the effects of IGF-1 diminishes with age [105]. Given these associations and the vital importance of GH/IGF-1 in promoting musculoskeletal growth, this pathway presents an attractive therapeutic target in addressing age-related osteoporosis and sarcopenia. Animal studies support this notion, showing a reversal in sarcopenia in aged rodents in response to GH treatment via effects in muscle protein synthesis, mitochondrial function, and oxidative stress [106].
However, the use of recombinant human growth hormone (rhGH) to reverse musculoskeletal aging is highly contentious. Twenty-five years ago, a famous study by Rudman and colleagues in older men treated with rhGH for 6 months demonstrated improvements in lean mass (8.8 %) and lumbar bone density (1.6 %) but no change in femoral neck density [107]. Despite the small number of participants (n = 21) and the lack of a control group, this study provoked intense interest in the ‘anti-aging’ potential of rhGH. Subsequent studies and a recent meta-analysis have tempered this excitement by demonstrating inconsistent effects of rhGH on bone density and physical function in older adults and concerning side effects including diabetes, edema, arthralgias, and entrapment neuropathy [108–110]. These studies are also limited by variable dosing regimens of rhGH, short treatment duration and follow-up periods (~6 months), and a lack of long-term safety data. In addition, there is the theoretical possibility that rhGH may increase mortality. This is suggested by a greater incidence of cancer and cardiovascular disease in patients with acromegaly (GH-secretory pituitary adenoma). Conversely, reduced GH/IGF-1 signaling has been shown to prolong lifespan in worms, insects, and mice [111]. In humans, GH deficiency and resistance are also associated with advanced longevity [101], and within the same population, short individuals have a statistical advantage in longevity versus tall individuals [112].
In spite of equivocal benefits of rhGH on musculoskeletal mass, its reported side effects and concerns of increased mortality, anti-aging clinics across the US have spawned a lucrative industry in the off-label use of rhGH as an “elixir of youth”. This raises serious concerns. In vitro studies report increased proliferation of cancer cells in response to rhGH [113]. In mice, rhGH enhances the growth of hepatic carcinoma xenografts [114]. There is also a published report of an elderly male with Crohn’s disease who developed IGF-1 receptor-expressing metastatic colon cancer 7 years after commencing rhGH [115]. Therefore longer-term and larger clinical trials are needed to determine the risks and benefits of rhGH in older subjects together with functional outcomes in osteoporosis and sarcopenia.
In principle, GH secretagogues are a more physiological alternative to rhGH. GH secretagogues result in pulsatile rather than prolonged increases in GH and respond to negative feedback by IGF-1. There have been small studies of GH secretagogues (GHRH-1,44-amide and ghrelin mimetic MK-677) which have demonstrated improvements in lean mass, unclear effects in physical function, and no change in bone mineral density [116, 117]. The largest randomized clinical trial of a GH secretagogue (capromorelin) involved 395 elderly subjects and had a planned duration of 2 years [118]. The trial was ceased prematurely due to significant increases in weight gain (1.4 kg at 6 months), arising from the appetite-stimulatory effect of this drug’s ghrelin mimetic action. On a positive note, capromorelin was associated with improvements in lean mass, tandem walking, and stair climbing [118]. However, subjects in this trial had mild functional decline and it therefore remains unclear whether GH secretagogues may have similar functional effects or improvements in bone density in frail subjects. There are also a number of downstream targets of GH/IGF-1, such as SOCS proteins, Grb10 [119], and IGF binding proteins (IGFBP) that may present future therapeutic targets which could potentially circumvent undesirable systemic effects of GH therapy.
Androgens and Selective Androgen Receptor Modulators (SARMs)
In addition to established effects in the reproductive system, androgens regulate growth by exerting anabolic effects in the musculoskeletal system. These effects are complex, relying on the activation of the androgen receptor (AR) at these sites, and the accrual of muscle tissue via an increase in protein synthesis, muscle fiber hypertrophy, and an increase in satellite cell number [120]. In bone, androgen-mediated increases in cortical and trabecular mass rely on the local conversion of testosterone to estrogen and the activation of estrogen receptors (ER) in osteoblasts [121].
Muscle wasting and osteoporosis are cardinal features of male hypogonadism and are amenable to testosterone therapy [122]. Similarly, men with muscle wasting due to HIV or chronic glucocorticoid therapy display increased muscle mass and strength in response to testosterone therapy [123]. The potential for testosterone therapy to reverse musculoskeletal aging has been suggested by a number of observations. Elderly men with reduced serum testosterone have a higher risk of muscle and bone loss and low-energy fracture [124]. Following testosterone therapy, older men display significant increases in bone density and lean mass [125, 126]. Studies suggest that testosterone therapy leads to improvements in strength in older men, related to muscle fiber type-specific effects and increased muscle mass [126, 127]. However, effects in bone were limited to the lumbar spine and the incidence of hard outcomes such as falls/fractures in those receiving testosterone therapy have not been established.
Safety concerns have been raised regarding the long-term use of testosterone therapy in older subjects. In particular, cardiovascular events and prostate cancer have been raised as potential risks, generating significant controversy in this field. In 2010, a study of community-dwelling older men was terminated due to a significantly higher incidence of cardiovascular incidents following the use of transdermal testosterone gel [128]. Larger studies have failed to clearly demonstrate the greater risk of cardiovascular events and in a statement this year, the FDA reported that the evidence was inconclusive [129]. However, a large meta-analysis of non-industry funded trials and a US-based epidemiologic study showed a two-fold increase in the risk of cardiovascular events with testosterone [130, 131]. Despite these concerns, prescription sales for testosterone in the US increased by 25 % each year from 1993 to 2002 [132].
Tissue-specific agents that exploit anabolic effects of androgens in muscle/bone, while circumventing troublesome effects in other tissues, have been studied for decades [133]. Selective androgen receptor modulators (SARMs) achieve tissue selectivity by differences in tissue distribution of AR isoforms and local interactions with 5-alpha reductase and aromatase [123]. Non-steroidal SARMs (e.g., quinolones, aryl propionamides) have greater oral bioavailability, AR specificity, and tissue selectivity than steroidal SARMs (e.g., 19-nortestosterone, 17-alpha-methyl-testosterone) and their development has therefore progressed further. Andarine (also known as S-4) is considered the ideal SARM due to its high oral bioavailability, long biological half-life allowing once-daily dosing, and preclinical data demonstrating consistent anabolic effects in muscle and bone [134]. A related compound, Ostarine (GTx-024, enobosarm), has been studied in phase I–III clinical trials and shown to increase lean mass and physical function in elderly men, post-menopausal women, and cancer patients [135–137]. Effects in bone density, however, have not been demonstrated possibly due to relatively short study periods of up to 3 months [135]. The agent LGD-4033 increased muscle mass and strength in healthy males in 3 weeks [138], and according to the company website (www.ligand.com), had beneficial effects in bone mass in preclinical studies. A phase II trial for this agent is planned for disorders of muscle wasting (e.g., cancer, fracture). In a recent mouse study, GLPG0492 reduced muscle fiber atrophy in immobilized muscles by modulating atrophy and tissue pleiotropic pathways involving MuRF1, myogenin, and FOXO1 [139]. Other agents such as BMS-564929 and LGD-2941 have shown similar results and are currently in phase I trials for age-related functional decline.
SARMs, therefore, hold great promise as anabolic agents that may reverse effects of musculoskeletal aging and by their tissue selectivity, circumvent concerns arising from off-target effects of AR activation. However, functional outcomes and long-term side effects of these agents need to be further explored.
Exercise and Nutrition
For decades, we have been instructing our patients with osteoporosis to participate in weight-bearing exercise. Enhanced muscle tone and balance, reduced falls, and bone loss, particularly of the femoral neck, are beneficial effects of regular exercise [140, 141]. Mechanical forces activate diverse signaling pathways involved in bone formation and muscle protein turnover, thereby forming a plausible basis for the beneficial effects of regular exercise [142]. Indeed, a recent extension study demonstrated long-term benefits of exercise in improving gait, parameters of physical function, and reducing falls in older individuals [143]. However these effects rely on continued participation in exercise. Positive effects in muscle strength and bone density were lost within 6 months of ceasing regular exercise in a group of post-menopausal women [144]. There is no consensus on the type, duration, or intensity of exercise that is most effective. Some studies advocate the benefits of regular walking [140] while others suggest music-based programs and resistance exercises [143, 145].
Malnutrition is rife in the elderly, affecting up to 40 % of those living in institutions [146]. It is also estimated that 1 in 5 older individuals in the USA consumes inadequate amounts of protein (i.e., <0.66 g/kg body weight per day) [147]. Malnutrition may exacerbate musculoskeletal aging as nutrients form substrates that are essential for muscle and bone accretion (protein, calcium, magnesium, phosphate) and dietary protein correlates with muscle mass and fractures [139]. However, protein supplements have not shown consistent effects in improving muscle mass and strength [148]. Small studies have suggested some benefit of essential amino acid (EAA) and leucine supplementation in improving muscle function in community-dwelling older individuals [149]. However, these studies were highly variable in terms of quality, supplemental doses, and the inclusion of exercise, and therefore cannot guide practice.
The use of calcium supplements for bone health is contentious. Calcium supplements lead to small increments in bone mineral density but these effects do not persist beyond their duration of use and they do not reduce fractures [150]. On the other hand, meta-analyses have linked calcium supplements with an increased risk of myocardial infarction, raising serious concerns about their widespread use [151, 152]. In general, it appears that potential risks of calcium supplementation, particularly in the elderly and those with renal impairment, outweigh their minimal benefits.
For 25 years, we have known of the association between vitamin K deficiency and osteoporotic fractures [143]. Vitamin K2 carboxylates osteocalcin, facilitating the incorporation of calcium into hydroxyapatite, increases the production of other matrix Gla proteins and may inhibit osteoclast-mediated bone resorption [145, 153]. However, RCTs of vitamin K supplementation have not shown significant improvements in bone mineral density or fracture risk among the elderly [154]. The use of vitamin K supplements for this group therefore remains an open question.
In summary, exercise and nutritional interventions lead to equivocal and short-lived results in muscle/bone mass in older individuals. The current evidence base is limited by small studies of variable quality and interventional strategies. Larger, standardized trials examining clinically meaningful outcomes in response to exercise/dietary intervention are needed before guidelines can be considered.
Future Directions
In addition to myostatin, there are myriad muscle-secreted factors, known as myokines, that also affect bone and may serve as therapeutic targets in musculoskeletal disorders. These include FGF-2, IL-6, IGF-1, BDNF, FSTL-1, and irisin [33]. As an example, IL6 stimulates bone resorption and IL6-related cytokines, Oncostatin M, and ciliary neurotrophic factor (CNTF), alters osteoblast differentiation and bone strength [155, 156]. Bone-secreted factors may also present important therapeutic targets. Osteocalcin alters muscle mitochondrial function [157] and lower limb strength in older women [36]. Sclerostin regulates the osteogenic response to muscle loading and is already being targeted for treatment in osteoporosis [158], although its effects in muscle mass are unclear. Increased understanding of the interconnected nature of the musculoskeletal system and the vast network of osteokines and myokines will potentially open new avenues of research in bone/muscle therapeutics.
Conclusions
Musculoskeletal aging, defined by the concurrent atrophy of muscle and bone, the infiltration of adipose tissue at these sites, and the attenuation of muscle fibers and bone mineral, is a highly complex and integrated process. Functionally, this progressive involution of muscle and bone leads to postural imbalance, deconditioning, falls and ultimately, fractures with dire consequences on morbidity and mortality. Our current efforts in reducing fracture are geared heavily toward bone and neglect the vastly intricate network of signals that govern and connect bone and muscle mass. An integrated approach that targets the muscle–bone unit as a whole maybe more effective in “breaking the vicious cycle” of musculoskeletal aging by exerting concurrent effects that reduce falls and fractures.
There has been significant progress in the development of novel anabolic therapies for muscle and bone, most notably activin pathway inhibitors, SARMs, and non-calcemic VDR agonists. Preclinical data show clear musculoskeletal benefits of these agents but the demonstration of unequivocal effects in falls and fractures are yet to be seen. This will depend on phase III clinical trials which are yet to be conducted for the majority of these therapies.
There are major challenges in the development of dual muscle–bone therapies. The definition of sarcopenia remains unclear and functional endpoints and markers of the ‘musculoskeletal unit’ are needed to confirm positive outcomes in efficacy trials. There have also been safety concerns with these agents and these relate to off-target effects of manipulating widespread signaling pathways. Activin pathway inhibitors have been hindered by reports of telangiectasia, bleeding, and gonadotropin suppression [90]. Similar issues have been overcome for therapies targeting other pathways. To circumvent undesirable effects of androgens, SARMs have been developed to specifically target muscle and bone. GH secretagogues preserve IGF-1-mediated negative feedback of GH and avoid the effects of supraphysiological levels of GH due to GH therapy [123]. Finally, VDR agonists may specifically target muscle and bone without dose-limiting systemic effects on calcium and phosphate homeostasis.
In recognizing the integrated biology of the musculoskeletal system and the complex signals involved in bone/muscle aging, the time has come to reconsider our approach to the prevention of osteoporotic fracture. Collaborative efforts by basic scientists, physicians, and the pharmaceutical industry are needed to address these complicated issues and ignite the development of novel therapies for the combined treatment of osteoporosis and sarcopenia.
References
Australian Bureau of Statistics Report on Musculoskeletal Conditions in Australia (2005) http://www.abs.gov.au/ausstats/abs@.nsf/cat/4823.0.55.001
Connelly L, Woolf A, Brooks P (2006) Cost-effectiveness of interventions for musculoskeletal conditions. In: Disease control in developing countries. Bank W, Washington DC
Johnell O, Kanis JA (2006) An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int 17:1726–1733
Liu-Ambrose T, Eng JJ, Khan KM, Carter ND, McKay HA (2003) Older women with osteoporosis have increased postural sway and weaker quadriceps strength than counterparts with normal bone mass: overlooked determinants of fracture risk? J Gerontol A Biol Sci Med Sci 58:M862–M866
Bonewald LF, Kiel DP, Clemens TL, Esser K, Orwoll ES, O’Keefe RJ, Fielding RA (2013) Forum on bone and skeletal muscle interactions: summary of the proceedings of an ASBMR workshop. J Bone Miner Res 28:1857–1865
Sjoblom S, Suuronen J, Rikkonen T, Honkanen R, Kroger H, Sirola J (2013) Relationship between postmenopausal osteoporosis and the components of clinical sarcopenia. Maturitas 75:175–180
Hida T, Ishiguro N, Shimokata H, Sakai Y, Matsui Y, Takemura M, Terabe Y, Harada A (2013) High prevalence of sarcopenia and reduced leg muscle mass in Japanese patients immediately after a hip fracture. Geriatr Gerontol Int 13:413–420
Binkley N, Krueger D, Buehring B (2013) What’s in a name revisited: should osteoporosis and sarcopenia be considered components of “dysmobility syndrome?”. Osteoporos Int 24:2955–2959
Cooper C, Dere W, Evans W, Kanis JA, Rizzoli R, Sayer AA, Sieber CC, Kaufman JM, Abellan van Kan G, Boonen S, Adachi J, Mitlak B, Tsouderos Y, Rolland Y, Reginster JY (2012) Frailty and sarcopenia: definitions and outcome parameters. Osteoporos Int 23:1839–1848
Cederholm T, Morley JE (2015) Sarcopenia: the new definitions. Curr Opin Clin Nutr Metab Care 18:1–4
Scott D, Hayes A, Sanders KM, Aitken D, Ebeling PR, Jones G (2014) Operational definitions of sarcopenia and their associations with 5-year changes in falls risk in community-dwelling middle-aged and older adults. Osteoporos Int 25:187–193
Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, Martin FC, Michel JP, Rolland Y, Schneider SM, Topinkova E, Vandewoude M, Zamboni M (2010) Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 39:412–423
Baumgartner RN, Koehler KM, Gallagher D, Romero L, Heymsfield SB, Ross RR, Garry PJ, Lindeman RD (1998) Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 147:755–763
DiGirolamo DJ, Kiel DP, Esser KA (2013) Bone and skeletal muscle: neighbors with close ties. J Bone Miner Res 28:1509–1518
DiGirolamo DJ, Kiel DP, Esser KA (2013) Bone and skeletal muscle: neighbors with close ties. J Bone Miner Res 28:1509–1518
Schoenwolf GC, Brauer PR, Francis-West PH (2009) Chapter 8: Development of the musculoskeletal system. In: Schoenwolf GC, LarsenWJ (eds) Larsen’s human embryology, 4th edn. Churchill Livingstone/Elsevier, Philadelphia
Karasik D, Kiel DP (2010) Evidence for pleiotropic factors in genetics of the musculoskeletal system. Bone 46:1226–1237
Larson CM, Henderson RC (2000) Bone mineral density and fractures in boys with Duchenne muscular dystrophy. J Pediatr Orthop 20:71–74
Shaw NJ, White CP, Fraser WD, Rosenbloom L (1994) Osteopenia in cerebral palsy. Arch Dis Child 71:235–238
Kahn J, Shwartz Y, Blitz E, Krief S, Sharir A, Breitel DA, Rattenbach R, Relaix F, Maire P, Rountree RB, Kingsley DM, Zelzer E (2009) Muscle contraction is necessary to maintain joint progenitor cell fate. Dev Cell 16:734–743
Nowlan NC, Bourdon C, Dumas G, Tajbakhsh S, Prendergast PJ, Murphy P (2010) Developing bones are differentially affected by compromised skeletal muscle formation. Bone 46:1275–1285
Sharir A, Stern T, Rot C, Shahar R, Zelzer E (2011) Muscle force regulates bone shaping for optimal load-bearing capacity during embryogenesis. Development 138:3247–3259
Slizewski A, Schonau E, Shaw C, Harvati K (2013) Muscle area estimation from cortical bone. Anat Rec (Hoboken) 296:1695–1707
Szulc P, Beck TJ, Marchand F, Delmas PD (2005) Low skeletal muscle mass is associated with poor structural parameters of bone and impaired balance in elderly men—the MINOS study. J Bone Miner Res 20:721–729
Rikkonen T, Sirola J, Salovaara K, Tuppurainen M, Jurvelin JS, Honkanen R, Kroger H (2012) Muscle strength and body composition are clinical indicators of osteoporosis. Calcif Tissue Int 91:131–138
Marcotte GR, West DW, Baar K (2014) The molecular basis for load-induced skeletal muscle hypertrophy. Calcif Tissue Int. doi:10.1007/s00223-014-9925-9
Keyak JH, Koyama AK, LeBlanc A, Lu Y, Lang TF (2009) Reduction in proximal femoral strength due to long-duration spaceflight. Bone 44:449–453
Shahnazari M, Wronski T, Chu V, Williams A, Leeper A, Stolina M, Ke HZ, Halloran B (2012) Early response of bone marrow osteoprogenitors to skeletal unloading and sclerostin antibody. Calcif Tissue Int 91:50–58
Moustafa A, Sugiyama T, Prasad J, Zaman G, Gross TS, Lanyon LE, Price JS (2012) Mechanical loading-related changes in osteocyte sclerostin expression in mice are more closely associated with the subsequent osteogenic response than the peak strains engendered. Osteoporos Int 23:1225–1234
Duncan RL, Turner CH (1995) Mechanotransduction and the functional response of bone to mechanical strain. Calcif Tissue Int 57:344–358
Lebrasseur NK, Achenbach SJ, Melton LJ 3rd, Amin S, Khosla S (2012) Skeletal muscle mass is associated with bone geometry and microstructure and serum insulin-like growth factor binding protein-2 levels in adult women and men. J Bone Miner Res 27:2159–2169
Elkasrawy MN, Hamrick MW (2010) Myostatin (GDF-8) as a key factor linking muscle mass and bone structure. J Musculoskelet Neuronal Interact 10:56–63
Pedersen BK, Febbraio MA (2012) Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 8:457–465
Cianferotti L, Brandi ML (2013) Muscle-bone interactions: basic and clinical aspects. Endocrine
DiGirolamo DJ, Clemens TL, Kousteni S (2012) The skeleton as an endocrine organ. Nat Rev Rheumatol 8:674–683
Levinger I, Scott D, Nicholson GC, Stuart AL, Duque G, McCorquodale T, Herrmann M, Ebeling PR, Sanders KM (2014) Undercarboxylated osteocalcin, muscle strength and indices of bone health in older women. Bone 64C:8–12
Christoforidis A, Maniadaki I, Stanhope R (2005) Growth hormone/insulin-like growth factor-1 axis during puberty. Pediatr Endocrinol Rev 3:5–10
Harry LE, Sandison A, Paleolog EM, Hansen U, Pearse MF, Nanchahal J (2008) Comparison of the healing of open tibial fractures covered with either muscle or fasciocutaneous tissue in a murine model. J Orthop Res 26:1238–1244
Gopal S, Majumder S, Batchelor AG, Knight SL, De Boer P, Smith RM (2000) Fix and flap: the radical orthopaedic and plastic treatment of severe open fractures of the tibia. J Bone Joint Surg Br 82:959–966
Reverte MM, Dimitriou R, Kanakaris NK, Giannoudis PV (2011) What is the effect of compartment syndrome and fasciotomies on fracture healing in tibial fractures? Injury 42:1402–1407
Rosen CJ, Adams JS, Bikle DD, Black DM, Demay MB, Manson JE, Murad MH, Kovacs CS (2012) The nonskeletal effects of vitamin D: an endocrine society scientific statement. Endocr Rev 33:456–492
Whistler D Thesis dissertation: De morbo puerili Anglorum quem patrio idiomate indigenae vocant ‘The Rickets’, 1645 University of Leyden, Leyden
Girgis CM, Clifton-Bligh RJ, Hamrick MW, Holick MF, Gunton JE (2013) The roles of vitamin D in skeletal muscle: form, function, and metabolism. Endocr Rev 34:33–83
Amling M, Priemel M, Holzmann T, Chapin K, Rueger JM, Baron R, Demay MB (1999) Rescue of the skeletal phenotype of vitamin D receptor-ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology 140:4982–4987
Schubert L, DeLuca HF (2010) Hypophosphatemia is responsible for skeletal muscle weakness of vitamin D deficiency. Arch Biochem Biophys 500:157–161
Johnson JA, Grande JP, Roche PC, Kumar R (1996) Ontogeny of the 1,25-dihydroxyvitamin D3 receptor in fetal rat bone. J Bone Miner Res 11:56–61
van Driel M, van Leeuwen JP (2014) Vitamin D endocrine system and osteoblasts. Bonekey Rep 3:493
Pojednic RM, Ceglia L, Olsson K, Gustafsson T, Lichtenstein AH, Dawson-Hughes B, Fielding RA (2014) Effects of 1,25-dihydroxy vitamin D and vitamin D on the expression of the vitamin D receptor in human skeletal muscle cells. Calcif Tissue Int. doi:10.1007/s00223-014-9932-x
Girgis CM, Mokbel N, Minn Cha K, Houweling PJ, Abboud M, Fraser DR, Mason RS, Clifton-Bligh RJ, Gunton JE (2014) The vitamin D receptor (VDR) is expressed in skeletal muscle of male mice and modulates 25-hydroxyvitamin D (25OHD) uptake in myofibers. Endocrinology 155:3227–3237
Girgis CM, Clifton-Bligh RJ, Mokbel N, Cheng K, Gunton JE (2014) Vitamin D signaling regulates proliferation, differentiation, and myotube size in C2C12 skeletal muscle cells. Endocrinology 155:347–357
Ceglia L, Niramitmahapanya S, Morais MD, Rivas DA, Harris SS, Bischoff-Ferrari H, Fielding RA, Dawson-Hughes B (2013) A randomized study on the effect of vitamin D3 supplementation on skeletal muscle morphology and vitamin D receptor concentration in older women. J Clin Endocrinol Metab 98:E1927–E1935
Lee SG, Lee YH, Kim KJ, Lee W, Kwon OH, Kim JH (2013) Additive association of vitamin D insufficiency and sarcopenia with low femoral bone mineral density in noninstitutionalized elderly population: the Korea National Health and Nutrition Examination Surveys 2009-2010. Osteoporos Int 24:2789–2799
Snijder MB, van Schoor NM, Pluijm SM, van Dam RM, Visser M, Lips P (2006) Vitamin D status in relation to one-year risk of recurrent falling in older men and women. J Clin Endocrinol Metab 91:2980–2985
Girgis CM (2014) Vitamin D and muscle function in the elderly: the elixir of youth? Curr Opin Clin Nutr Metab Care 17:546–550
Bischoff-Ferrari HA, Borchers M, Gudat F, Durmuller U, Stahelin HB, Dick W (2004) Vitamin D receptor expression in human muscle tissue decreases with age. J Bone Miner Res 19:265–269
Bischoff-Ferrari HA, Willett WC, Wong JB, Giovannucci E, Dietrich T, Dawson-Hughes B (2005) Fracture prevention with vitamin D supplementation: a meta-analysis of randomized controlled trials. JAMA 293:2257–2264
Broe KE, Chen TC, Weinberg J, Bischoff-Ferrari HA, Holick MF, Kiel DP (2007) A higher dose of vitamin D reduces the risk of falls in nursing home residents: a randomized, multiple-dose study. J Am Geriatr Soc 55:234–239
Jackson RD, LaCroix AZ, Gass M, Wallace RB, Robbins J, Lewis CE, Bassford T, Beresford SA, Black HR, Blanchette P, Bonds DE, Brunner RL, Brzyski RG, Caan B, Cauley JA, Chlebowski RT, Cummings SR, Granek I, Hays J, Heiss G, Hendrix SL, Howard BV, Hsia J, Hubbell FA, Johnson KC, Judd H, Kotchen JM, Kuller LH, Langer RD, Lasser NL, Limacher MC, Ludlam S, Manson JE, Margolis KL, McGowan J, Ockene JK, O’Sullivan MJ, Phillips L, Prentice RL, Sarto GE, Stefanick ML, Van Horn L, Wactawski-Wende J, Whitlock E, Anderson GL, Assaf AR, Barad D (2006) Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med 354:669–683
Reid IR, Bolland MJ, Grey A (2013) Effects of vitamin D supplements on bone mineral density: a systematic review and meta-analysis. Lancet 383:146–155
Bleicher K, Cumming RG, Naganathan V, Blyth FM, Le Couteur DG, Handelsman DJ, Waite LM, Seibel MJ (2014) U-shaped association between serum 25-hydroxyvitamin D and fracture risk in older men: results from the prospective population based CHAMP study. J Bone Miner Res 29:2024–2031. doi:10.1002/jbmr.2230
Ensrud KE, Ewing SK, Fredman L, Hochberg MC, Cauley JA, Hillier TA, Cummings SR, Yaffe K, Cawthon PM (2010) Circulating 25-hydroxyvitamin D levels and frailty status in older women. J Clin Endocrinol Metab 95:5266–5273
Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, Durazo-Arvizu RA, Gallagher JC, Gallo RL, Jones G, Kovacs CS, Mayne ST, Rosen CJ, Shapses SA (2011) The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 96:53–58
Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, Murad MH, Weaver CM (2011) Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 96:1911–1930
Sanders KM, Stuart AL, Williamson EJ, Simpson JA, Kotowicz MA, Young D, Nicholson GC (2010) Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA 303:1815–1822
Iwamoto J, Sato Y (2014) Eldecalcitol improves chair-rising time in postmenopausal osteoporotic women treated with bisphosphonates. Ther Clin Risk Manag 10:51–59
Matsumoto T, Ito M, Hayashi Y, Hirota T, Tanigawara Y, Sone T, Fukunaga M, Shiraki M, Nakamura T (2011) A new active vitamin D3 analog, eldecalcitol, prevents the risk of osteoporotic fractures—a randomized, active comparator, double-blind study. Bone 49:605–612
Takeda S, Smith SY, Tamura T, Saito H, Takahashi F, Samadfam R, Haile S, Doyle N, Endo K (2015) Long-term treatment with eldecalcitol (1alpha, 25-dihydroxy-2beta- (3-hydroxypropyloxy) vitamin D) suppresses bone turnover and leads to prevention of bone loss and bone fragility in ovariectomized rats. Calcif Tissue Int 96:45–55
Sakai S, Suzuki M, Tashiro Y, Tanaka K, Takeda S, Aizawa K, Hirata M, Yogo K, Endo K (2015) Vitamin D receptor signaling enhances locomotive ability in mice. J Bone Miner Res 30:128–136
McPherron AC, Lawler AM, Lee SJ (1997) Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387:83–90
Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ (2004) Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350:2682–2688
Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M, Nair G, Arver S, Bhasin S (1998) Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci USA 95:14938–14943
Han DS, Chen YM, Lin SY, Chang HH, Huang TM, Chi YC, Yang WS (2011) Serum myostatin levels and grip strength in normal subjects and patients on maintenance haemodialysis. Clin Endocrinol (Oxf) 75:857–863
Ju CR, Chen RC (2012) Serum myostatin levels and skeletal muscle wasting in chronic obstructive pulmonary disease. Respir Med 106:102–108
Zhang ZL, He JW, Qin YJ, Hu YQ, Li M, Zhang H, Hu WW, Liu YJ, Gu JM (2008) Association between myostatin gene polymorphisms and peak BMD variation in Chinese nuclear families. Osteoporos Int 19:39–47
Hamrick MW (2003) Increased bone mineral density in the femora of GDF8 knockout mice. Anat Rec A Discov Mol Cell Evol Biol 272:388–391
Kellum E, Starr H, Arounleut P, Immel D, Fulzele S, Wenger K, Hamrick MW (2009) Myostatin (GDF-8) deficiency increases fracture callus size, Sox-5 expression, and callus bone volume. Bone 44:17–23
Elliott B, Renshaw D, Getting S, Mackenzie R (2012) The central role of myostatin in skeletal muscle and whole body homeostasis. Acta Physiol (Oxf) 205:324–340
Tsuchida K, Nakatani M, Hitachi K, Uezumi A, Sunada Y, Ageta H, Inokuchi K (2009) Activin signaling as an emerging target for therapeutic interventions. Cell Commun Signal 7:15
Digirolamo D, Singhal V, Clemens T, Lee S-J (2011) Systemic administration of soluble 483 activin receptors produces differential anabolic effects in muscle and bone in mice. J Bone Miner Res (Suppl.):1167
Bowser M, Herberg S, Arounleut P, Shi X, Fulzele S, Hill WD, Isales CM, Hamrick MW (2013) Effects of the activin A-myostatin-follistatin system on aging bone and muscle progenitor cells. Exp Gerontol 48:290–297
Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ (2010) Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142:531–543
Chiu CS, Peekhaus N, Weber H, Adamski S, Murray EM, Zhang HZ, Zhao JZ, Ernst R, Lineberger J, Huang L, Hampton R, Arnold BA, Vitelli S, Hamuro L, Wang WR, Wei N, Dillon GM, Miao J, Alves SE, Glantschnig H, Wang F, Wilkinson HA (2013) Increased muscle force production and bone mineral density in ActRIIB-Fc-treated mature rodents. J Gerontol A Biol Sci Med Sci 68:1181–1192
Koncarevic A, Cornwall-Brady M, Pullen A, Davies M, Sako D, Liu J, Kumar R, Tomkinson K, Baker T, Umiker B, Monnell T, Grinberg AV, Liharska K, Underwood KW, Ucran JA, Howard E, Barberio J, Spaits M, Pearsall S, Seehra J, Lachey J (2010) A soluble activin receptor type IIb prevents the effects of androgen deprivation on body composition and bone health. Endocrinology 151:4289–4300
Lotinun S, Pearsall RS, Davies MV, Marvell TH, Monnell TE, Ucran J, Fajardo RJ, Kumar R, Underwood KW, Seehra J, Bouxsein ML, Baron R (2010) A soluble activin receptor type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone 46:1082–1088
Arounleut P, Bialek P, Liang LF, Upadhyay S, Fulzele S, Johnson M, Elsalanty M, Isales CM, Hamrick MW (2013) A myostatin inhibitor (propeptide-Fc) increases muscle mass and muscle fiber size in aged mice but does not increase bone density or bone strength. Exp Gerontol 48:898–904
Bialek P, Parkington J, Li X, Gavin D, Wallace C, Zhang J, Root A, Yan G, Warner L, Seeherman HJ, Yaworsky PJ (2014) A myostatin and activin decoy receptor enhances bone formation in mice. Bone 60:162–171
Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, Flanigan KM, Pestronk A, Tawil R, Wolfe GI, Eagle M, Florence JM, King WM, Pandya S, Straub V, Juneau P, Meyers K, Csimma C, Araujo T, Allen R, Parsons SA, Wozney JM, Lavallie ER, Mendell JR (2008) A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 63:561–571
Krivickas LS, Walsh R, Amato AA (2009) Single muscle fiber contractile properties in adults with muscular dystrophy treated with MYO-029. Muscle Nerve 39:3–9
Padhi D, Higano CS, Shore ND, Sieber P, Rasmussen E, Smith MR (2014) Pharmacological inhibition of myostatin and changes in lean body mass and lower extremity muscle size in patients receiving androgen deprivation therapy for prostate cancer. J Clin Endocrinol Metab 99:E1967–E1975
Attie KM, Borgstein NG, Yang Y, Condon CH, Wilson DM, Pearsall AE, Kumar R, Willins DA, Seehra JS, Sherman ML (2013) A single ascending-dose study of muscle regulator ACE-031 in healthy volunteers. Muscle Nerve 47:416–423
Ruckle J, Jacobs M, Kramer W, Pearsall AE, Kumar R, Underwood KW, Seehra J, Yang Y, Condon CH, Sherman ML (2009) Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J Bone Miner Res 24:744–752
Perrini S, Carreira MC, Conserva A, Laviola L, Giorgino F (2008) Metabolic implications of growth hormone therapy. J Endocrinol Invest 31:79–84
Perrini S, Laviola L, Carreira MC, Cignarelli A, Natalicchio A, Giorgino F (2010) The GH/IGF1 axis and signaling pathways in the muscle and bone: mechanisms underlying age-related skeletal muscle wasting and osteoporosis. J Endocrinol 205:201–210
Mavalli MD, DiGirolamo DJ, Fan Y, Riddle RC, Campbell KS, van Groen T, Frank SJ, Sperling MA, Esser KA, Bamman MM, Clemens TL (2010) Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J Clin Invest 120:4007–4020
Miller WL, Eberhardt NL (1983) Structure and evolution of the growth hormone gene family. Endocr Rev 4:97–130
Carter-Su C, Schwartz J, Smit LS (1996) Molecular mechanism of growth hormone action. Annu Rev Physiol 58:187–207
Giustina A, Mazziotti G, Canalis E (2008) Growth hormone, insulin-like growth factors, and the skeleton. Endocr Rev 29:535–559
Hill PA, Tumber A, Meikle MC (1997) Multiple extracellular signals promote osteoblast survival and apoptosis. Endocrinology 138:3849–3858
Hock JM, Centrella M, Canalis E (1988) Insulin-like growth factor I has independent effects on bone matrix formation and cell replication. Endocrinology 122:254–260
DiGirolamo DJ, Mukherjee A, Fulzele K, Gan Y, Cao X, Frank SJ, Clemens TL (2007) Mode of growth hormone action in osteoblasts. J Biol Chem 282:31666–31674
Laron Z (2005) Do deficiencies in growth hormone and insulin-like growth factor-1 (IGF-1) shorten or prolong longevity? Mech Ageing Dev 126:305–307
Zhao HY, Liu JM, Ning G, Zhao YJ, Chen Y, Sun LH, Zhang LZ, Xu MY, Chen JL (2008) Relationships between insulin-like growth factor-I (IGF-I) and OPG, RANKL, bone mineral density in healthy Chinese women. Osteoporos Int 19:221–226
Leger B, Derave W, De Bock K, Hespel P, Russell AP (2008) Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res 11:163B–175B
Rivas DA, Lessard SJ, Rice NP, Lustgarten MS, So K, Goodyear LJ, Parnell LD, Fielding RA (2014) Diminished skeletal muscle microRNA expression with aging is associated with attenuated muscle plasticity and inhibition of IGF-1 signaling. Faseb J 28:4133–4147
Ghiron LJ, Thompson JL, Holloway L, Hintz RL, Butterfield GE, Hoffman AR, Marcus R (1995) Effects of recombinant insulin-like growth factor-I and growth hormone on bone turnover in elderly women. J Bone Miner Res 10:1844–1852
Brioche T, Kireev RA, Cuesta S, Gratas-Delamarche A, Tresguerres JA, Gomez-Cabrera MC, Vina J (2014) Growth hormone replacement therapy prevents sarcopenia by a dual mechanism: improvement of protein balance and of antioxidant defenses. J Gerontol A Biol Sci Med Sci 69:1186–1198
Rudman D, Feller AG, Nagraj HS, Gergans GA, Lalitha PY, Goldberg AF, Schlenker RA, Cohn L, Rudman IW, Mattson DE (1990) Effects of human growth hormone in men over 60 years old. N Engl J Med 323:1–6
Liu H, Bravata DM, Olkin I, Nayak S, Roberts B, Garber AM, Hoffman AR (2007) Systematic review: the safety and efficacy of growth hormone in the healthy elderly. Ann Intern Med 146:104–115
Papadakis MA, Grady D, Black D, Tierney MJ, Gooding GA, Schambelan M, Grunfeld C (1996) Growth hormone replacement in healthy older men improves body composition but not functional ability. Ann Intern Med 124:708–716
Blackman MR, Sorkin JD, Munzer T, Bellantoni MF, Busby-Whitehead J, Stevens TE, Jayme J, O’Connor KG, Christmas C, Tobin JD, Stewart KJ, Cottrell E, St Clair C, Pabst KM, Harman SM (2002) Growth hormone and sex steroid administration in healthy aged women and men: a randomized controlled trial. JAMA 288:2282–2292
Berryman DE, Christiansen JS, Johannsson G, Thorner MO, Kopchick JJ (2008) Role of the GH/IGF-1 axis in lifespan and healthspan: lessons from animal models. Growth Horm IGF Res 18:455–471
T S (2007) Human body size and the laws of scaling: physiological, performance, growth, longevity and ecological ramifications. Nova Science Publishers, New York
Li S, Hou G, Wang Y, Su X, Xue L (2010) Influence of recombinant human growth hormone (rhGH) on proliferation of hepatocellular carcinoma cells with positive and negative growth hormone receptors in vitro. Tumori 96:282–288
Liu JP, Chen T, Ling YB, Chen XX, Ou QJ (2006) Effect of recombinant human growth hormone on growth of human Bel-7402 hepatic carcinoma xenografts in nude mice. Ai Zheng 25:292–296
Melmed GY, Devlin SM, Vlotides G, Dhall D, Ross S, Yu R, Melmed S (2008) Anti-aging therapy with human growth hormone associated with metastatic colon cancer in a patient with Crohn’s colitis. Clin Gastroenterol Hepatol 6:360–363
Veldhuis JD, Patrie JM, Frick K, Weltman JY, Weltman AL (2005) Administration of recombinant human GHRH-1,44-amide for 3 months reduces abdominal visceral fat mass and increases physical performance measures in postmenopausal women. Eur J Endocrinol 153:669–677
Nass R, Pezzoli SS, Oliveri MC, Patrie JT, Harrell FE Jr, Clasey JL, Heymsfield SB, Bach MA, Vance ML, Thorner MO (2008) Effects of an oral ghrelin mimetic on body composition and clinical outcomes in healthy older adults: a randomized trial. Ann Intern Med 149:601–611
White HK, Petrie CD, Landschulz W, MacLean D, Taylor A, Lyles K, Wei JY, Hoffman AR, Salvatori R, Ettinger MP, Morey MC, Blackman MR, Merriam GR (2009) Effects of an oral growth hormone secretagogue in older adults. J Clin Endocrinol Metab 94:1198–1206
Mokbel N, Hoffman NJ, Girgis CM, Small L, Turner N, Daly RJ, Cooney GJ, Holt LJ (2014) Grb10 deletion enhances muscle cell proliferation, differentiation and GLUT4 plasma membrane translocation. J Cell Physiol 229:1753–1764
Chen Y, Zajac JD, MacLean HE (2005) Androgen regulation of satellite cell function. J Endocrinol 186:21–31
Vanderschueren D, Laurent MR, Claessens F, Gielen E, Lagerquist MK, Vandenput L, Borjesson AE, Ohlsson C (2014) Sex steroid actions in male bone. Endocr Rev 35:906–960
Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, Montori VM (2010) Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 95:2536–2559
Bhasin S, Calof OM, Storer TW, Lee ML, Mazer NA, Jasuja R, Montori VM, Gao W, Dalton JT (2006) Drug insight: testosterone and selective androgen receptor modulators as anabolic therapies for chronic illness and aging. Nat Clin Pract Endocrinol Metab 2:146–159
Fink HA, Ewing SK, Ensrud KE, Barrett-Connor E, Taylor BC, Cauley JA, Orwoll ES (2006) Association of testosterone and estradiol deficiency with osteoporosis and rapid bone loss in older men. J Clin Endocrinol Metab 91:3908–3915
Tracz MJ, Sideras K, Bolona ER, Haddad RM, Kennedy CC, Uraga MV, Caples SM, Erwin PJ, Montori VM (2006) Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab 91:2011–2016
Isidori AM, Giannetta E, Greco EA, Gianfrilli D, Bonifacio V, Isidori A, Lenzi A, Fabbri A (2005) Effects of testosterone on body composition, bone metabolism and serum lipid profile in middle-aged men: a meta-analysis. Clin Endocrinol (Oxf) 63:280–293
Fitts RH, Peters JR, Dillon EL, Durham WJ, Sheffield-Moore M, Urban RJ (2014) Weekly versus monthly testosterone administration on fast and slow skeletal muscle fibers in older adult males. J Clin Endocrinol Metab. doi:10.1210/jc.2014-2759
Basaria S, Coviello AD, Travison TG, Storer TW, Farwell WR, Jette AM, Eder R, Tennstedt S, Ulloor J, Zhang A, Choong K, Lakshman KM, Mazer NA, Miciek R, Krasnoff J, Elmi A, Knapp PE, Brooks B, Appleman E, Aggarwal S, Bhasin G, Hede-Brierley L, Bhatia A, Collins L, LeBrasseur N, Fiore LD, Bhasin S (2010) Adverse events associated with testosterone administration. N Engl J Med 363:109–122
http://www.fda.gov/Drugs/DrugSafety/ucm383904.htm. (2014) FDA evaluating risk of stroke, heart attack and death with FDA-approved testosterone products
Xu L, Freeman G, Cowling BJ, Schooling CM (2013) Testosterone therapy and cardiovascular events among men: a systematic review and meta-analysis of placebo-controlled randomized trials. BMC Med 11:108
Finkle WD, Greenland S, Ridgeway GK, Adams JL, Frasco MA, Cook MB, Fraumeni JF Jr, Hoover RN (2014) Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PLoS One 9:e85805
Liverman CT (eds) (2004) Testosterone and aging: clinical research directions. Institute of Medicine TNAP, Washington, DC
Zhang X, Sui Z (2013) Deciphering the selective androgen receptor modulators paradigm. Expert Opin Drug Discov 8:191–218
Mohler ML, Bohl CE, Jones A, Coss CC, Narayanan R, He Y, Hwang DJ, Dalton JT, Miller DD (2009) Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit. J Med Chem 52:3597–3617
Dalton JT, Barnette KG, Bohl CE, Hancock ML, Rodriguez D, Dodson ST, Morton RA, Steiner MS (2011) The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: results of a double-blind, placebo-controlled phase II trial. J Cachexia Sarcopenia Muscle 2:153–161
Dobs AS, Boccia RV, Croot CC, Gabrail NY, Dalton JT, Hancock ML, Johnston MA, Steiner MS (2013) Effects of enobosarm on muscle wasting and physical function in patients with cancer: a double-blind, randomised controlled phase 2 trial. Lancet Oncol 14:335–345
Bristow SM, Gamble GD, Stewart A, Horne L, House ME, Aati O, Mihov B, Horne AM, Reid IR (2014) Acute and 3-month effects of microcrystalline hydroxyapatite, calcium citrate and calcium carbonate on serum calcium and markers of bone turnover: a randomised controlled trial in postmenopausal women. Br J Nutr 112:1611–1620
Basaria S, Collins L, Dillon EL, Orwoll K, Storer TW, Miciek R, Ulloor J, Zhang A, Eder R, Zientek H, Gordon G, Kazmi S, Sheffield-Moore M, Bhasin S (2013) The safety, pharmacokinetics, and effects of LGD-4033, a novel nonsteroidal oral, selective androgen receptor modulator, in healthy young men. J Gerontol A Biol Sci Med Sci 68:87–95
Calvani R, Martone AM, Marzetti E, Onder G, Savera G, Lorenzi M, Serafini E, Bernabei R, Landi F (2014) Pre-hospital dietary intake correlates with muscle mass at the time of fracture in older hip-fractured patients. Front Aging Neurosci 6:269
Korpelainen R, Keinanen-Kiukaanniemi S, Heikkinen J, Vaananen K, Korpelainen J (2006) Effect of impact exercise on bone mineral density in elderly women with low BMD: a population-based randomized controlled 30-month intervention. Osteoporos Int 17:109–118
Martyn-St James M, Carroll S (2008) Meta-analysis of walking for preservation of bone mineral density in postmenopausal women. Bone 43:521–531
Gilsanz V, Wren TA, Sanchez M, Dorey F, Judex S, Rubin C (2006) Low-level, high-frequency mechanical signals enhance musculoskeletal development of young women with low BMD. J Bone Miner Res 21:1464–1474
Bitensky L, Hart JP, Catterall A, Hodges SJ, Pilkington MJ, Chayen J (1988) Circulating vitamin K levels in patients with fractures. J Bone Joint Surg Br 70:663–664
Winters KM, Snow CM (2000) Detraining reverses positive effects of exercise on the musculoskeletal system in premenopausal women. J Bone Miner Res 15:2495–2503
Bugel S (2003) Vitamin K and bone health. Proc Nutr Soc 62:839–843
Serrano-Urrea R, Garcia-Meseguer MJ (2013) Malnutrition in an elderly population without cognitive impairment living in nursing homes in Spain: study of prevalence using the Mini Nutritional Assessment Test. Gerontology 59:490–498
Berner LA, Becker G, Wise M, Doi J (2013) Characterization of dietary protein among older adults in the United States: amount, animal sources, and meal patterns. J Acad Nutr Diet 113:809–815
Milne AC, Potter J, Vivanti A, Avenell A (2009) Protein and energy supplementation in elderly people at risk from malnutrition. Cochrane Database Syst Rev. doi:10.1002/14651858.CD003288.pub3
Cruz-Jentoft AJ, Landi F, Schneider SM, Zuniga C, Arai H, Boirie Y, Chen LK, Fielding RA, Martin FC, Michel JP, Sieber C, Stout JR, Studenski SA, Vellas B, Woo J, Zamboni M, Cederholm T (2014) Prevalence of and interventions for sarcopenia in ageing adults: a systematic review. Report of the International Sarcopenia Initiative (EWGSOP and IWGS). Age Ageing 43:748–759
Tang BM, Eslick GD, Nowson C, Smith C, Bensoussan A (2007) Use of calcium or calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50 years and older: a meta-analysis. Lancet 370:657–666
Radford LT, Bolland MJ, Mason B, Horne A, Gamble GD, Grey A, Reid IR (2013) The Auckland calcium study: 5-year post-trial follow-up. Osteoporos Int
Reid IR, Bristow SM, Bolland MJ (2014) Cardiovascular Complications of Calcium Supplements. J Cell Biochem. doi:10.1002/jcb.25028
Yamaguchi M, Uchiyama S, Tsukamoto Y (2003) Inhibitory effect of menaquinone-7 (vitamin K2) on the bone-resorbing factors-induced bone resorption in elderly female rat femoral tissues in vitro. Mol Cell Biochem 245:115–120
Hamidi MS, Cheung AM (2014) Vitamin K and musculoskeletal health in postmenopausal women. Mol Nutr Food Res 58:1647–1657
Johnson RW, Brennan HJ, Vrahnas C, Poulton IJ, McGregor NE, Standal T, Walker EC, Koh TT, Nguyen H, Walsh NC, Forwood MR, Martin TJ, Sims NA (2013) The primary function of gp130 signaling in osteoblasts is to maintain bone formation and strength, rather than promote osteoclast formation. J Bone Miner Res 29:1492–1505
Johnson RW, White JD, Walker EC, Martin TJ, Sims NA (2014) Myokines (muscle-derived cytokines and chemokines) including ciliary neurotrophic factor (CNTF) inhibit osteoblast differentiation. Bone 64C:47–56
Clemens TL, Karsenty G (2011) The osteoblast: an insulin target cell controlling glucose homeostasis. J Bone Miner Res 26:677–680
McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A, Langdahl BL, Reginster JY, Zanchetta JR, Wasserman SM, Katz L, Maddox J, Yang YC, Libanati C, Bone HG (2014) Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 370:412–420
Acknowledgments
CMG is funded by a National Health and Medical Research Council (NHMRC) Peter Doherty Early Career Research Fellowship. I also wish to acknowledge the kind assistance of A/Professor Jane Bleasel (Department of Rheumatology, Royal Prince Alfred Hospital, Sydney) for critically reviewing this manuscript.
Conflict of interest
Christian M. Girgis has no disclosures to report.
Human and Animal Rights and Informed Consent
All research discussed in this review has been subject to ethics approval and informed consent has been obtained in human studies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Girgis, C.M. Integrated Therapies for Osteoporosis and Sarcopenia: From Signaling Pathways to Clinical Trials. Calcif Tissue Int 96, 243–255 (2015). https://doi.org/10.1007/s00223-015-9956-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-015-9956-x