
Abstract
The skeleton is no longer seen as a static, isolated, and mostly structural organ. Over the last two decades, a more complete picture of the multiple functions of the skeleton has emerged, and its interactions with a growing number of apparently unrelated organs have become evident. The skeleton not only reacts to mechanical loading and inflammatory, hormonal, and mineral challenges, but also acts of its own accord by secreting factors controlling the function of other tissues, including the kidney and possibly the pancreas and gonads. It is thus becoming widely recognized that it is by nature an endocrine organ, in addition to a structural organ and site of mineral storage and hematopoiesis. Consequently and by definition, bone homeostasis must be tightly regulated and integrated with the biology of other organs to maintain whole body homeostasis, and data uncovering the involvement of the central nervous system (CNS) in the control of bone remodeling support this concept. The sympathetic nervous system (SNS) represents one of the main links between the CNS and the skeleton, based on a number of anatomic, pharmacologic, and genetic studies focused on β-adrenergic receptor (βAR) signaling in bone cells. The goal of this report was to review the data supporting the role of the SNS and βAR signaling in the regulation of skeletal homeostasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Skeleton Innervation
Discovery
Though the functional consequences of bone innervation, such as bone pain from fractures, gout, or arthritis, were documented in ancient Egypt [1], it was not until 1846 that Gros [2] demonstrated a large nerve entering the cortical bone via the nutrient foramen in equine, bovine, and human femorae. Since then, many studies demonstrated the presence of osseous nerve fibers, but there has been a dearth of knowledge regarding regional innervation, as well as specific types of fibers and functional relevance of these fibers to skeletal biology, mostly due to technical challenges involved in visualizing delicate nerve fibers within a calcified tissue and loss of antigenicity with decalcification procedures [3, 4]. Initially, a variety of silver and gold stains (using modifications of Cajal’s stain) were applied to visualize osseous innervation at a general structural level and to differentiate myelinated and unmyelinated fibers [5]. More recent immunofluorescence techniques revealed specific types of nerve fibers via the identification of specific sensory or sympathetic neuropeptides (NPs), such as neurofilament 200, nerve growth factor receptor (Trk-A), protein gene product 9.5 (PGP9.5), vasoactive intestinal peptide, substance P (SP), neuropeptide Y (NPY), within the different compartments of the bone [6, 7].
Bone is innervated according to Hilton’s rule, meaning the nervous supply of the overlying muscle and skin is continuous with the long bones and joints. Large nerve bundles accompany the major arteries that feed the bone at multiple locations on a long bone. The largest nerve enters the diaphysis via the nutrient foramen and innervates the medullary space. Others enter the bone at the articular extremities on both sides of the epiphyses.
Nerves that enter the bone marrow space are thought to be primarily sympathetic vasomotor, as evidenced by the presence of spiraling nervi vasorum around bone marrow blood vessels and numerous nervous plexi in the tunica media of bone arteries [8]. These nerves are positive for tyrosine hydroxylase, dopamine β-hydroxylase (DBH), and NPY. Sensory nerves are also present in vertebral bodies [9] as well as long bones [10]. Sensory fibers are detected in the bone marrow, though their function within the marrow space is unclear [9].
Periosteal and cortical bones are richly innervated, though it is unclear whether nerve fibers in the periosteum penetrate fully to the marrow. Periosteal innervation primarily consists of an extremely dense network of sensory fibers (positive for calcitonin gene-related peptide [CGRP] and SP) that are sensitive to both mechanical stimulation [11] and pain [10], though sympathetic fibers are also present in this bone compartment [12]. Many of these nerves penetrate into the cortical bone alongside the ligamentous Sharpey’s fibers, suggesting further sensory perception within the cortical bone, though this has not been demonstrated.
The overall pattern of nerve distribution within the bone microenvironment of a single bone element suggests that only a limited number of bone cells are in direct contact with nerve terminals [13, 14]. Therefore, signal transduction by neurotransmitters and NPs in bone cells could be nonsynaptic (such as norepinephrine [NE] spillover from vascular nerves) and/or could involve communication via intercellular junctions. The fact that osteoblasts and -cytes both express β2-adrenergic receptor (β2AR), one of the main receptors for NE released by sympathetic neurons and communicate through GAP junctions, supports this hypothesis.
Ontogenesis of Bone Innervation
The innervation of long bones plays a direct role in bone development, as evidenced from increased diaphyseal length in peroneal denervation experiments with juvenile rabbits more than 50 years ago [15]. During embryological development, the earliest GAP43-expressing nerve fibers appear at e17 in rats, followed closely by nerves expressing PGP9.5 (e19). PGP9.5- and GAP-positive nerves are first seen in rodent embryos in the perichondrium and -osteum of the limb buds, followed by infiltration of the diaphyseal marrow alongside blood vessels. Postnatally, expression of NPY, DBH, and vesicular acetylcholine transporter (VAChT) can be seen in periosteal and diaphyseal nerves with later expression (p7–p14) found in the secondary ossification centers and epiphyseal marrow space [16–19]. Afferent sensory innervation of bone during development has also been detailed. SP-expressing sensory nerves have been described in numerous mammalian species, including rodents, horses, and humans [18, 19]. Additionally, neurons have been observed in developing limb buds of birds and reptiles, indicating an important evolutionarily conserved function for nerves in the skeleton [20].
Semaphorins, which mediate axonal guidance, regulate the innervation of bone, specifically by Sema3A expression in bone cells [21]. This may represent a mechanism by which developing bones control their own innervation. Other important bone proteins such as BMP also dictate the extent of peripheral innervation of organs [22], though its role in the innervation of bone specifically has not been shown.
In addition to nerve growth seen in developing limbs, rapid sprouting of new CGRP sensory nerves correlates with the formation of new bone in deer antlers [23]. Bone formation in these animals is decreased after denervation [24, 25], suggesting a functional role for sensory innervation of bone growth in this model, though it is complicated by the dense blood vasculature necessary for antler formation. Rapid expansion of a dense network of CGRP-positive nerves is also observed during fracture repair in the periosteum of adult rodents [26–28], though the functional consequences are unclear and the nerves are not associated with blood vessels [29]. Developmental studies of bone innervation thus remain relatively incomplete, and the analysis of transgenic mice characterized by fluorescent nerve-specific markers and genetic mutant mice characterized by defect of bone innervation should in the near future contribute to a better characterization of skeletal innervation.
The Route from CNS to Bones
Anatomical links between the skeleton and specific centers in the central nervous system (CNS) show a wide range of connections. Retrograde tracing experiments using pseudorabies virus revealed spinal connections to the femur at the level of the dorsal root ganglia between L2 and L5 [30]. Other studies have further demonstrated that these neural connections to the bone extend far beyond the spine and into dozens of specific regions of the CNS, including brain stem, paraventricular nucleus of hypothalamus, prelimbic cortex, and motor cortex [13], implying a much deeper relationship between bone and the CNS than is currently recognized. It remains unclear whether skeletal elements formed by intramembranous versus endochondral bone formation display similar innervation or not, and whether differences of innervation between axial and appendicular bones exist.
βAR Signaling in Osteoblasts
Expression
Upon action potential, sympathetic nerves release NE and thereby activate postsynaptic βARs. There are three βAR: β1-, β2-, and β3ARs. Effects of adrenal extracts (containing the catecholamines NE and epinephrine) on bone growth were first noted in the 1950s [31], followed by observations of increased cyclic AMP (cAMP) upon epinephrine or isoproterenol treatment of bone marrow cells in the 1970s [32–35]. However, it took another decade for researchers to propose the functional relevance of βAR stimulation specifically in osteoblastic cells, when comparing PTH and calcitonin-induced increases in cAMP during in vitro studies using UMR 106 rat osteosarcoma cells [36]. βAR (specifically β2AR) expression was later confirmed in various cell lines (ROS 17/2.8, SaOS-2, HOS, MG-63) and mouse calvarial primary osteoblasts and shown to mediate profound effects on bone turnover [14, 37–41]. β2AR expression was also detected in osteoclast cultures, although data interpretation was complicated by the presence of osteoblasts and other β2AR-expressing cells in these cultures [42, 43]. In another study of β2AR signaling in osteoclasts, a ROS-mediated effect was seen in vitro [43]. However, biological evidence demonstrating the causal relationship between functional β2AR signaling in osteoclasts and bone resorption in vivo, independent of osteoblastic β2AR, is lacking since similar ROS data was seen in osteoblastic cells [44]. β2AR expression has also been found in chondrocytes [34, 45–48] and nerves have been documented in human cartilaginous epiphyses [27], providing the possibility of adrenergic effects on the growth plate. Additionally, a growth inhibitory effect of clenbuterol has been seen in rats [49], though these data contrast with the normal size of β2AR-deficient mice.
Unlike β2AR, expression of either β1- or β3ARs is less readily detected in primary osteoblasts or osteoblast cell lines [14, 37–39]. Two studies did detect β1AR expression in human and rat osteosarcoma cell lines [39, 50], and one study detected β3AR transcripts in osteosarcoma cell lines and rat long bone primary osteoblasts [51].
Signaling
β2ARs are seven-transmembrane G protein-coupled receptors (GPCRs) that primarily signal through Gs, a G protein which also couples to many other GPCRs that are important to bone homeostasis, such as PTHR, calcitonin receptor, PGE2, and 5HT2, and many others. The β2AR can also signal through Gi (Gs–Gi switch) and the beta/gamma (β/γ) subunit. The alpha (α) subunit of Gs allosterically modulates and activates adenylyl cyclase, leading to a Mg-dependent conversion of ATP to the second messenger cAMP. The accumulated intracellular cAMP binds regulatory units of protein kinase A (PKA) holoenzyme, which causes release of the catalytic PKA subunits. The freed catalytic portions can then phosphorylate serine and threonine residues of downstream proteins, such as those that compose calcium channels in the cell membrane, enzymes in the cytoplasm, and nuclear factors such as CREB or ATF4. Intracellular signal termination is mediated via the intrinsic GTPase activity of Gsα, which leads to the hydrolysis of GTP to GDP and the reassociation of Gsα-GDP and Gβγ subunits.
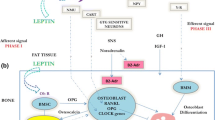
There are a number of signaling components known to be involved downstream of the β2AR that might contribute to β2AR signaling in osteoblasts, although experimental evidence for a direct link to β2AR signaling are not always available (Fig. 1). For instance, tight control of Gsα activity is required for normal skeletal homeostasis, as shown by the dramatic anabolic response of a mutant mouse model characterized by constitutive Gsα activity in osteoblasts [52]. Transduction by Gs-dependent GPCRs like the β2AR can be modulated by cAMP-specific phosphodiesterases (PDEs), which regulate the localization, duration, and amplitude of downstream signaling by decreasing cAMP concentrations via cleavage of cAMP to 5′-AMP. While PDE4 has been reported to increase Rankl expression in osteoblasts [53], others have shown that inhibition of PDE4 has the same effect [54]. In vivo, a PDE4 inhibitor, Rolipram, increased bone mass in mice, through osteoblast differentiation and bone formation, without affecting resorption parameters [55], but another study showed that it increased osteoclast formation [56]. The exact mechanisms and receptors controlled by PDEs in bone cells are thus still relatively uncharacterized, and it is at this point difficult to know whether it controls β2AR signaling in osteoblasts. β2AR signaling can also be attenuated via targeted receptor phosphorylation by G protein-coupled receptor kinases (GRK). Originally dubbed beta adrenergic receptor kinases (βARK), GRK proteins contribute to desensitization, internalization and recycling of desensitized receptors, independent of cAMP [57]. In the short term, GRKs decrease receptor signaling by rapidly phosphorylating specific residues of the GPCR to facilitate recruitment and binding of β-arrestins to the phosphorylated receptor, which decreases both agonist affinity and G protein binding by steric mechanisms [58]. In the long term, desensitization is facilitated by internalization of the GPCR, where it is either dephosphorylated or degraded. GRK2 and β-arrestin-1 are expressed in primary cultures of rat osteoblasts, UMR 106-H5 and ROS 17/2.8 osteoblastic cells [59, 60] and may play a role in regulating growth-factor responsiveness in osteoblasts [59]. Inhibiting GRK activity in mature osteoblasts by overexpression of a GRK inhibitor decreased bone remodeling and was anabolic [61]. On the other hand, overexpression of GRK2 increased bone remodeling and led to bone loss in young mice [62]. Although these findings support a significant role of GRK in mature osteoblasts and in attenuating GPCR desensitization, they do not support a model by which GRK activity controls β2AR signaling in osteoblasts, at least in the setting of the control of bone remodeling.

The β2AR is the central mediator of SNS signaling in osteoblasts. (a) Activation of the SNS releases catecholamines in the bone which activate osteoblastic β2AR. (b) Signaling is primarily mediated by the stimulatory G-alpha subunit (Gs), which activates adenylyl cyclase, resulting in increased intracellular cAMP, which then activates protein kinase A. (c) The active PKA catalytic subunits can then phosphorylate cytosolic proteins to increase intracellular calcium or translocate to the nucleus (d), where they activate CREB and ATF4. (e) These transcription factors modulate the expression of various genes within the osteoblast that affect its own function as well as the behavior of other cells, e.g., osteoclasts, within the bone. Multiple proteins such as arrestins, phosphodiesterases, and GPCR kinases have been shown to modulate β2AR transduction at different steps within the osteoblast. Additionally, other neurotransmitter receptors that signal through Gq likely also interact with β2AR, signaling transduction within the nucleus. Gi-coupled receptors may be involved in controlling the magnitude of β2AR effects along with other Gs-coupled receptors. (f) β2AR signaling in bone is also controlled centrally by signaling through brain-expressed adrenergic, cannabinoid, and muscarinic receptors, whose activation decreases SNS outflow
In conclusion, critical components of the cytoplasmic machinery used by GPCRs have been identified and shown to be functionally relevant in osteoblast; however, signaling and pathway interaction studies remain scarce and difficult to follow up in vivo due to the important temporal gap between these rapid cellular events and the processes they control in vivo.
Effects
Daily βAR stimulation, by means of the nonselective β1-/β2ARs agonist isoproterenol or the β2AR-selective agonists clenbuterol or salbutamol, in mice and rats triggers an osteoclastogenic response, as measured by bone loss and increased osteoclast formation [14, 63, 64]. This osteoclastogenic effect is thought to be mostly caused by an increase in RANKL and IL6 expression, which are both target genes of β2AR signaling in osteoblasts [41, 65]. β2AR expression in osteoblasts remains high upon in vitro differentiation. Osteocytes have recently been shown to be one of the major biologically relevant cells that provide RANKL to control bone remodeling in vivo [66, 67]. Although it remains unclear which types of osteoblasts (progenitor cells, active osteoblasts or fully differentiated osteoblasts/-cytes) are most responsive to NE or pharmacological βAR agonists, one can speculate that osteocytic RANKL production may be under the control of sympathetic nerves. It is also worth noting that immune cells, including T cells, express the β2AR and RANKL, and hence the immune compartment in bone may also represent a significant target of sympathetic nerves.
βAR stimulation also affects bone formation by inhibiting osteoblast proliferation, thus uncoupling osteoblasts and -clasts in vivo, leading to bone loss. Although the main molecular Clock gene machinery regulating body rhythms is located in the CNS, β2AR stimulation was found to signal via CREB to activate the circadian genes Per1 and Per2 in osteoblasts [68]. Osteoblastic Clock genes then inhibit c-myc transcription, leading to down-regulation of its target cyclin D1. β2AR stimulation in osteoblasts also activates AP-1 gene expression, which belongs to a family of genes known to regulate proliferation, in part through activation of c-myc and cyclin D1, thereby counteracting the Clock pathway.
Cumulative genetic evidence strongly supports the biological relevance of β2AR signaling in the regulation of bone remodeling. First, lack of Dopamine hydroxylase (DBH), an enzyme required for the synthesis of catecholamines, causes a high bone mass phenotype in 6-month-old mice [14]. Second, β2AR-deficient mice also display a high bone mass phenotype detectable by 3–4 months of age [41, 69]. In contrast to other mouse models characterized by low sympathetic tone and high cancellous bone mass, such as the ob/ob mice, β2AR-deficient mice do not present with obesity or gonadal dysfunction, thus reinforcing the hypothesis that β2AR signaling in osteoblasts contributes to the regulation of bone homeostasis. The observation that osteoblast-specific inactivation of the β2AR (using the floxed allele of Adrβ2 and the 2.3 kb α1Col1 Cre-deleter mice) induces a high bone mass phenotype confirmed the osteoblast-specific role of the β2AR in the regulation of bone remodeling and supported the notion that sympathetic signals, mainly via the β2AR expressed in osteoblasts, restrain bone formation and favors bone resorption [70].
Although the β2AR appears to be the main βAR involved in the regulation of bone remodeling in osteoblasts, global genetic inactivation of the β1AR also had significant consequences on bone, as measured by a decreased bone mass in β1AR-deficient and β1-/β2ARs-deficient mice [71, 72]. The bone phenotype of β1-/β2ARs-deficient mice was accompanied by reduced body weight and low serum IGF1 level (Table 1) [72]. In addition, β1AR stimulation by dobutamine partly protected rats from the deleterious effect of unloading on bone formation and strength [73]. On the other hand, global β1-/β2-/β3ARs genetic inactivation led to a high bone mass phenotype, accompanied by increased body weight and, accordingly, increased leptin levels (Table 1) [74]. Considering the low levels of β1- and β3ARs transcripts in the osteoblast lineage, the changes in body weight, the abnormal hormonal status of double or triple KO mice (Table 1) and the bone phenotype of mice lacking brown adipose tissue (where the β3AR is highly expressed) [75], these results suggest that adrenergic signaling affects skeleton homeostasis through direct effects on bone and indirect effects via other nerve-targeted tissues.
The aforementioned findings suggested that pharmacological βAR antagonism may have a beneficial effect on bone. This hypothesis has been investigated in rodents, mainly through the use of propranolol, a nonselective β1-/β2ARs antagonist. These studies overall showed that propranolol, like β2AR genetic ablation, has a bone anabolic effect, with a more pronounced effect under conditions such as ovariectomy [14, 69, 76], mechanical unloading [77] or hypertension [78–80]. Each of these conditions exhibit increased osteoclast number and bone resorption, supporting the hypothesis that propranolol may be more efficient in reducing a state of increased bone resorption than in attenuating the basal rate of bone resorption. In addition, low dose propranolol was demonstrated to have a better bone mass preservative effect in rats than high dose, and this was associated with increased IGF1 serum levels [69, 76, 78]. Whether this is true in mice remains unknown. On the basis of the low bone mass of the β1AR- and β1-/β2ARs-deficient mice, one possible explanation for this differential dose effect may be that low dose propranolol might primarily block the β2AR in osteoblasts, whereas high dose propranolol may antagonize other βARs expressed in bone cells or other tissues. Alternatively, the benefits of low dose over high dose propranolol could be due to off-target autonomic effects of β-blockers [81] and/or AR or signaling supersensitization, as is the case reported with some patients on β-blockers [82–84], or sensitization of other signaling pathways significant to osteoblasts [85–87].
If sympathetic outflow and βAR signaling are intrinsic components of the bone remodeling process, one can speculate that conditions affecting these systems may have impact on the skeleton. A first illustration of this idea is the effect of dexamethasone on the expression level of β2AR. Glucocorticoids indeed were shown to stimulate β2AR expression in differentiated osteoblasts and were proposed to enhance the responsiveness of these cells to sympathetic stimulation, hence leading to bone loss [88]. Such effects may be relevant to low bone mass conditions associated with increased endogenous or high-dose exogenous glucocorticoids. Another interesting corroboration linking sympathetic outflow, βAR signaling and bone remodeling is the association between osteoporosis and severe depression [89–94], which might be in part explained by the increase in sympathetic outflow caused by depression [95–97], as supported by data in a mouse model of depression [98]. Lastly, recent evidence suggested that the effect of sympathetic nerves on the skeleton might not be limited to the regulation of bone remodeling. Sympathetic activation induced by chronic stress/depression indeed was shown to create, in mice, a bone microenvironment favorable for the establishment of metastatic breast cancer cells, acting in part by up-regulating the expression and release of RANKL, a cytokine promoting osteoclastogenesis but also breast cancer cell migration [99]. Although not directly tested, this effect may be directly related to clinical evidence showing an association between depression and shorter survival in women with breast cancer [100–103]. In addition, mouse data suggest that βAR stimulation in osteoblasts decreases insulin levels in mice, thus linking the activity of nerves in bones and osteoblasts to the regulation of glucose homeostasis [104].
α-AR Signaling and Bone Remodeling: Central or Peripheral Action?
There are two αAR subtypes: α1- (a Gq coupled receptor) and α2ARs (a Gi coupled receptor). α1ARs are primarily located in smooth muscles and contribute to smooth muscle constriction. α2ARs (α2A, α2B, α2C) are expressed mainly in the pancreas and in presynaptic adrenergic neurons, and play a key role in regulating neurotransmitter release in the CNS and peripheral sympathetic nervous system (SNS). Activation of the α2ARs in the brain stem leads to a reduction in sympathetic tone and a resultant decrease in heart rate and blood pressure, whereas deletion of the α2AR leads to increased sympathetic outflow [105]. Stimulation of this receptor by endogenous NE is under the dependence of another regulatory mechanism involving NE reuptake by the NE transporter (NET), a monoamine transporter and a member of the Na+/Cl-dependent family of neurotransmitter transporters. The amount of endogenous NE released by sympathetic neurons is indeed controlled presynaptically by NET and the reuptake of NE into presynaptic neurons, which clears 80–90 % of NE released into synaptic clefts in the CNS and peripheral SNS. Whether these two important presynaptic central regulators of sympathetic outflow play a direct role in the control of bone remodeling is unknown and the focus of ongoing studies. There is, however, evidence for a possible bone cell-autonomous role of αARs. α2A and to a lesser extent α2B and α2CAR transcripts can be detected at low levels in bone, in osteoblasts, -cytes, -clasts and in MC3T3 cells [46]. α1A, α1B, and α1D mRNA can be detected in myeloid progenitors, calvaria osteoblasts, and MC3T3 cells as well [106–108]. However, evidence for a functional relevance of these receptors expressed in bone cells in the regulation of bone remodeling is still relatively limited, as in vivo studies published so far used a global αAR inactivation strategy. Genetic global deletion of α2A-/α2CARs caused a high bone mass phenotype in female mice, accompanied mainly by reduced bone resorption [46]. In vitro osteoclast differentiation experiments suggested that α2AR activation in osteoclasts promotes their differentiation [46], and in another study, doxazosin (an α-blocker) was observed to increase osteogenic differentiation of human mesenchymal stem cells in vitro [109]. The fact that α2AAR-deficient mice are characterized by high sympathetic tone and a high bone mass, rather than the expected low bone mass, suggests that the mechanisms whereby endogenous sympathetic tone regulate skeletal homeostasis are far more complicated than previously thought, and shed light on the importance of other possible mechanisms whereby catecholamines affect osteoblast and osteoclast biology. The analysis of α2AAR cell-specific conditional mutant mice will be necessary to understand the role of this receptor in bone homeostasis and to clarify its site of action.
The role of the cannabinoid receptors CB1 in the context of sympathetic outflow seems also clearly important in the control of bone homeostasis. CB1 is expressed in sympathetic terminals, and mouse studies suggest that 2-arachidonoylglycerol (2-AG) activation of prejunctional CB1 suppresses NE release from sympathetic terminals in bone, thus alleviating the inhibition of bone formation. Opposite phenotypes between mouse models characterized by CB1 deficiency, however, still obscure our understanding of the role of the endocannabinoid system and CB1 in bone remodeling [29, 110, 111].
In conclusion, although the role of postsynaptic β2AR signaling in bone remodeling has become well established, at least in preclinical models, most recent studies highlight the importance of αAR signaling and possible differences between β2AR stimulation by exogenous pharmacological drugs and endogenous catecholamines in the regulation of bone homeostasis.
Let’s Not Forget the Parasympathetic Nervous System
The autonomic nervous system is divided functionally and anatomically into two antagonistic arms, the SNS and parasympathetic nervous systems (PSNS). Therefore, a role of the PSNS in skeletal biology has been suspected for some time. Acetylcholine is the main neurotransmitter used by the PSNS. It is synthesized in presynaptic neurons by acetylation of choline and packaged in presynaptic vesicles through the action of the VAChT. It binds to the nicotinic ACh receptors (nAChR), which function primarily as ion channels in certain neurons and on postsynaptic cells of the neuromuscular junction. It also binds to seven-transmembrane domain muscarinic receptors (mAChR). The M1-, M3-, and M5Rs selectively couple to G proteins of the Gq family, while the M2- and M4Rs preferentially activate Gi/Go-type of G proteins. In postsynaptic cells, ACh esterase (AChE) degrades ACh, thus limiting its action.
Evidence supporting a role of the PSNS in skeletal homeostasis includes the correlation of cigarette smoking, and the related increase in nicotine levels (a cholinergic agonist), to higher fracture risk [112], as well as the detection of nicotinic and muscarinic receptors in osteoblasts [113–115]. More recently, genetic studies have shown that the PSNS, acting through the M3 muscarinic receptor, favors bone mass accrual by increasing bone formation and decreasing bone resorption [116]. This conclusion was drawn on the evidence that M 3 R deficiency (and not M 1 -, M 2 -, M 4 Rs deficiency) causes a low bone mass phenotype in mice. In addition, this study showed that mice lacking M3R in Nestin-positive cells (generated using the Nestin-Cre transgenic mice) have a low bone mass phenotype (whereas osteoblast-specific [α1(I)Collagen-Cre] M3R-deficient mice do not), and that ablating genetically one copy of the β2AR in M3R+/−; β2AR+/− mice rescued their bone phenotype. These studies thus suggested that the PSNS favors bone mass accrual by a central mode of action and by inhibiting sympathetic outflow. Parasympathetic innervation in bone was confirmed by detection of VAChT-positive nerve fibers in bone marrow close to trabeculae [115]. This study also used pseudorabies virus-based tracing studies to document a physical link between the appendicular skeleton (femur) and central autonomic nuclei. It also addressed, via the analysis of α2nAChR−/− mice, the role of nicotinic receptors in skeletal homeostasis, and showed that α2nAChR mainly regulates bone resorption, α2nAChR−/− mice displaying an osteoclast-driven low bone mass phenotype, and nAChR activation enhancing osteoclast apoptosis and inhibiting mineralized matrix resorption [115].
Why Does All This Matter?
There are obvious clinical implications to the thus far acquired sum of preclinical data in this new field of neuroskeletal biology. One is the potential effect of β-blockers on fracture risk or bone mineral density. Studies vary to a great extent in terms of patient number, drug exposure time, doses, AR selectivity, age of patients, menopausal status and methodologies, but overall β-blocker use has been mainly associated with reduced fracture risk and increased bone mineral density [117–121], although not all studies support this conclusion [122, 123]. These inconsistencies between clinical studies, beyond methodological and patient number issues, might be related to the opposite effects of β1 and β2AR inactivation on bone mass revealed in genetic mouse mutants, the distinct results of high versus low dose propranolol treatment shown in rats, and the preferential and more effective activity of propranolol in conditions associated with high bone turn over. Yet all these possible explanations remain speculative.
Another implication of these studies is a possible effect of autonomic disorders on bone. Patients with autonomic dysfunction (pure autonomic failure, DBH deficiency), who have a low sympathetic tone, are rare but sustain an impressive number of falls (ranging from 50 to 500 per year) in spite of being considerably more bedridden than the average age-matched population [124–127]. Repetitive falls and disuse osteoporosis in these patients, which derive from their orthostatic hypotension, should logically lead to increased fracture risk [128, 129]. However, this is not supported by any observations in the literature. This suggests that low sympathetic tone caused by autonomic dysfunctions in humans increases bone density, as observed in murine models. The opposite could be true for patients with postural tachycardia syndrome, who are characterized by increased sympathetic tone [130–132].
Conversely, β2AR stimulation has been associated with bone loss. Exposure to β2AR agonists in patients with asthma/COPD increased hip/femur fracture risk, but this effect was attenuated after exclusion of oral glucocorticoid users and adjustment for the underlying disease [133]. The fact that glucocorticoids increase β2AR expression in osteoblasts could be related to the effect of these adjustments [88]. In addition, pheochromocytoma and the related increase in catecholamine levels was shown to be associated with increased bone resorption, and adrenalectomy to normalize bone resorption blood parameters [134], further supporting the functional contribution of AR signaling in the regulation of skeletal homeostasis in humans.
The aforementioned preclinical data and retrospective clinical data suggest that increased sympathetic activity and reduced parasympathetic activity may contribute to osteoporosis, and thus that neurological risk factors may be important to the etiology of osteoporosis. A number of studies support this hypothesis, although all of them remain associative. A correlation between rapid resting heart rate and increased risk of osteoporotic fractures has been reported, for example [135]. In addition, increased sympathetic activity and reduced parasympathetic activity was measured in postmenopausal women with osteoporosis compared to postmenopausal women without osteoporosis, using heart rate variability parameters and sympathetic skin responses [136]. In another study, which compared sympathetic activity between pre- and postmenopausal women and using microneurography at the peroneal nerve, increased sympathetic activity was recorded in the post- compared to premenopausal group. In the two groups combined and after age adjustment, sympathetic activity was inversely correlated with trabecular bone volume fraction, thickness and compressive bone strength, as assessed by high resolution peripheral quantitative computed tomography and microfinite element (μFE) analysis [137].
In conclusion, the discovery that the SNS is an intrinsic component of the regulatory machinery controlling skeletal homeostasis unmasked the integration and multifunctionality of the skeleton within the whole body, and emphasized its endocrine nature. The relevance of these findings to clinical human pathophysiology is still a matter of debate, however, and regardless of their future validity and usefulness in the clinic, these studies are very interesting examples of integrative biology.
References
Ebell BB (1937) The Paprus Ebers: the greatest Egyptian medical document. Oxford University Press, London
Gros M (1846) La Disposition des nerfs des os. Bull Soc Anat Paris 21:369–372
Hayat M (2002) Factors affecting antigen retrieval. In: Hayat MA (ed) Microscopy, immunohistochemistry, and antigen retrieval methods for light and electron microscopy. Springer, pp 53–69
Shi SR, Key ME, Kalra KL (1991) Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem 39:741–748
de Castro F (1929) Quelques observations sur l’intervention du système nerveux autonome dans l’ossification. Innervation du tissu osseux et de la moelle osseuse. T. Travaux du Lab Rec Biol Univ Madrid 26:215
Castaneda-Corral G, Jimenez-Andrade JM, Bloom AP, Taylor RN, Mantyh WG, Kaczmarska MJ, Ghilardi JR, Mantyh PW (2011) The majority of myelinated and unmyelinated sensory nerve fibers that innervate bone express the tropomyosin receptor kinase A. Neuroscience 178:196–207
Bjurholm A, Kreicbergs A, Terenius L, Goldstein M, Schultzberg M (1988) Neuropeptide Y-, tyrosine hydroxylase- and vasoactive intestinal polypeptide-immunoreactive nerves in bone and surrounding tissues. J Auton Nerv Syst 25:119–125
Duncan CP, Shim SS (1977) J. Edouard Samson Address: the autonomic nerve supply of bone. An experimental study of the intraosseous adrenergic nervi vasorum in the rabbit. J Bone Jt Surg Br 59:323–330
Ohtori S, Inoue G, Koshi T, Ito T, Watanabe T, Yamashita M, Yamauchi K, Suzuki M, Doya H, Moriya H, Takahashi Y, Takahashi K (2007) Sensory innervation of lumbar vertebral bodies in rats. Spine 32:1498–1502
Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, Keyser CP, Clohisy DR, Adams DJ, O’Leary P, Mantyh PW (2002) Origins of skeletal pain: sensory and sympathetic innervation of the mouse femur. Neuroscience 113:155–166
Mahns DA, Ivanusic JJ, Sahai V, Rowe MJ (2006) An intact peripheral nerve preparation for monitoring the activity of single, periosteal afferent nerve fibres. J Neurosci Methods 156:140–144
Fan W, Bouwense SA, Crawford R, Xiao Y (2010) Structural and cellular features in metaphyseal and diaphyseal periosteum of osteoporotic rats. J Mol Histol 41:51–60
Denes A, Boldogkoi Z, Uhereczky G, Hornyak A, Rusvai M, Palkovits M, Kovacs KJ (2005) Central autonomic control of the bone marrow: multisynaptic tract tracing by recombinant pseudorabies virus. Neuroscience 134:947–963
Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, Armstrong D, Ducy P, Karsenty G (2002) Leptin regulates bone formation via the sympathetic nervous system. Cell 111:305–317
Ring PA (1961) The influence of the nervous system upon the growth of bones. J Bone Jt Surg Br 43:121–140
Gajda M, Adriaensen D, Cichocki T (2000) Development of the innervation of long bones: expression of the growth-associated protein 43. Folia Histochem Cytobiol 38:103–110
Gajda M, Litwin JA, Tabarowski Z, Zagolski O, Cichocki T, Timmermans JP, Adriaensen D (2010) Development of rat tibia innervation: colocalization of autonomic nerve fiber markers with growth-associated protein 43. Cells Tissues Organs 191:489–499
Sisask G, Bjurholm A, Ahmed M, Kreicbergs A (1995) Ontogeny of sensory nerves in the developing skeleton. Anat Rec 243:234–240
Sisask G, Bjurholm A, Ahmed M, Kreicbergs A (1996) The development of autonomic innervation in bone and joints of the rat. J Auton Nerv Syst 59:27–33
Martini R, Schachner M (1991) Complex expression pattern of tenascin during innervation of the posterior limb buds of the developing chicken. J Neurosci Res 28:261–279
Gomez C, Burt-Pichat B, Mallein-Gerin F, Merle B, Delmas PD, Skerry TM, Vico L, Malaval L, Chenu C (2005) Expression of Semaphorin-3A and its receptors in endochondral ossification: potential role in skeletal development and innervation. Dev Dyn 234:393–403
Guha U, Gomes WA, Samanta J, Gupta M, Rice FL, Kessler JA (2004) Target-derived BMP signaling limits sensory neuron number and the extent of peripheral innervation in vivo. Development 131:1175–1186
Gray C, Hukkanen M, Konttinen YT, Terenghi G, Arnett TR, Jones SJ, Burnstock G, Polak JM (1992) Rapid neural growth: calcitonin gene-related peptide and substance P-containing nerves attain exceptional growth rates in regenerating deer antler. Neuroscience 50:953–963
Singh IJ, Herskovits MS, Chiego DJ Jr, Klein RM (1982) Modulation of osteoblastic activity by sensory and autonomic innervation of bone. Prog Clin Biol Res 101:535–551
Suttie JM, Fennessy PF (1985) Regrowth of amputated velvet antlers with and without innervation. J Exp Zool 234:359–366
Hukkanen M, Konttinen YT, Santavirta S, Paavolainen P, Gu XH, Terenghi G, Polak JM (1993) Rapid proliferation of calcitonin gene-related peptide-immunoreactive nerves during healing of rat tibial fracture suggests neural involvement in bone growth and remodelling. Neuroscience 54:969–979
Strange-Vognsen HH, Laursen H (1997) Nerves in human epiphyseal uncalcified cartilage. J Pediatr Orthop B 6:56–58
Li J, Ahmad T, Spetea M, Ahmed M, Kreicbergs A (2001) Bone reinnervation after fracture: a study in the rat. J Bone Miner Res 16:1505–1510
Tam J, Trembovler V, Di Marzo V, Petrosino S, Leo G, Alexandrovich A, Regev E, Casap N, Shteyer A, Ledent C, Karsak M, Zimmer A, Mechoulam R, Yirmiya R, Shohami E, Bab I (2008) The cannabinoid CB1 receptor regulates bone formation by modulating adrenergic signaling. FASEB J 22:285–294
Edoff K, Grenegard M, Hildebrand C (2000) Retrograde tracing and neuropeptide immunohistochemistry of sensory neurones projecting to the cartilaginous distal femoral epiphysis of young rats. Cell Tissue Res 299:193–200
Maassen AP (1952) The influence of adrenalectomy on the growth of rats. Arch Int Pharmacodyn Ther 88:473–481
Paul MI, Kvetnansky R, Cramer H, Silbergeld S, Kopin IJ (1971) Immobilization stress induced changes in adrenocortical and medullary cyclic AMP content in the rat. Endocrinology 88:338–344
Smith DM, Johnston CC Jr (1974) Hormonal responsiveness of adenylate cyclase activity from separate bone cells. Endocrinology 95:130–139
Wong GL (1979) Induction of metabolic changes and down regulation of bovine parathyroid hormone-responsive adenylate cyclase are dissociable in isolated osteoclastic and osteoblastic bone cells. J Biol Chem 254:34–37
Lipski S (1976) Effects of beta-adrenergic stimulation on bone-marrow function in normal and sublethally irradiated mice. I. The effect of isoproterenol on cAMP content in bone-marrow cells in vivo and in vitro. Int J Radiat Biol Relat Stud Phys Chem Med 29:359–366
Gutierrez GE, Mundy GR, Katz MS (1984) Adenylate cyclase of osteoblast-like cells from rat osteosarcoma is stimulated by calcitonin as well as parathyroid hormone. Endocrinology 115:2342–2346
Moore RE, Smith CK II, Bailey CS, Voelkel EF, Tashjian AH Jr (1993) Characterization of beta-adrenergic receptors on rat and human osteoblast-like cells and demonstration that beta-receptor agonists can stimulate bone resorption in organ culture. Bone Miner 23:301–315
Togari A, Arai M, Mizutani S, Mizutani S, Koshihara Y, Nagatsu T (1997) Expression of mRNAs for neuropeptide receptors and beta-adrenergic receptors in human osteoblasts and human osteogenic sarcoma cells. Neurosci Lett 233:125–128
Kellenberger S, Muller K, Richener H, Bilbe G (1998) Formoterol and isoproterenol induce c-fos gene expression in osteoblast-like cells by activating beta2-adrenergic receptors. Bone 22:471–478
Majeska RJ, Minkowitz B, Bastian W, Einhorn TA (1992) Effects of beta-adrenergic blockade in an osteoblast-like cell line. J Orthop Res 10:379–384
Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, Liu X, Kondo H, Richards WG, Bannon TW, Noda M, Clement K, Vaisse C, Karsenty G (2005) Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature 434:514–520
Aitken SJ, Landao-Bassonga E, Ralston SH, Idris AI (2009) Beta2-adrenoreceptor ligands regulate osteoclast differentiation in vitro by direct and indirect mechanisms. Arch Biochem Biophys 482:96–103
Kondo H, Takeuchi S, Togari A (2013) Beta-adrenergic signaling stimulates osteoclastogenesis via reactive oxygen species. Am J Physiol Endocrinol Metab 304:E507–E515
Takahata Y, Takarada T, Iemata M, Yamamoto T, Nakamura Y, Kodama A, Yoneda Y (2009) Functional expression of beta2 adrenergic receptors responsible for protection against oxidative stress through promotion of glutathione synthesis after Nrf2 upregulation in undifferentiated mesenchymal C3H10T1/2 stem cells. J Cell Physiol 218:268–275
Lai LP, Mitchell J (2008) Beta2-adrenergic receptors expressed on murine chondrocytes stimulate cellular growth and inhibit the expression of Indian hedgehog and collagen type X. J Cell Biochem 104:545–553
Fonseca TL, Jorgetti V, Costa CC, Capelo LP, Covarrubias AE, Moulatlet AC, Teixeira MB, Hesse E, Morethson P, Beber EH, Freitas FR, Wang CC, Nonaka KO, Oliveira R, Casarini DE, Zorn TM, Brum PC, Gouveia CH (2011) Double disruption of alpha2A- and alpha2C-adrenoceptors results in sympathetic hyperactivity and high-bone-mass phenotype. J Bone Miner Res 26:591–603
Mitchell J, Lai LP, Peralta F, Xu Y, Sugamori K (2011) Beta2-adrenergic receptors inhibit the expression of collagen type II in growth plate chondrocytes by stimulating the AP-1 factor Jun-B. Am J Physiol Endocrinol Metab 300:E633–E639
Vignon E, Broquet P, Mathieu P, Louisot P, Richard M (1990) Histaminergic H1, serotoninergic, beta adrenergic and dopaminergic receptors in human osteoarthritic cartilage. Biochem Int 20:251–255
Kitaura T, Tsunekawa N, Kraemer WJ (2002) Inhibited longitudinal growth of bones in young male rats by clenbuterol. Med Sci Sports Exerc 34:267–273
Whitsett JA, Burdsall J, Workman L, Hollinger B, Neely J (1983) Beta-adrenergic receptors in pediatric tumors: uncoupled beta 1-adrenergic receptor in Ewing’s sarcoma. J Natl Cancer Inst 71:779–786
Nuntapornsak A, Wongdee K, Thongbunchoo J, Krishnamra N, Charoenphandhu N (2010) Changes in the mRNA expression of osteoblast-related genes in response to beta(3)-adrenergic agonist in UMR106 cells. Cell Biochem Funct 28:45–51
Hsiao EC, Boudignon BM, Chang WC, Bencsik M, Peng J, Nguyen TD, Manalac C, Halloran BP, Conklin BR, Nissenson RA (2008) Osteoblast expression of an engineered Gs-coupled receptor dramatically increases bone mass. Proc Natl Acad Sci USA 105:1209–1214
Park H, No AL, Lee JM, Chen L, Lee SY, Lee DS, Yim M (2010) PDE4 inhibitor upregulates PTH-induced osteoclast formation via CRE-mediated COX-2 expression in osteoblasts. FEBS Lett 584:173–180
Cho ES, Yu JH, Kim MS, Yim M (2004) Rolipram, a phosphodiesterase 4 inhibitor, stimulates osteoclast formation by inducing TRANCE expression in mouse calvarial cells. Arch Pharmacol Res 27:1258–1262
Kinoshita T, Kobayashi S, Ebara S, Yoshimura Y, Horiuchi H, Tsutsumimoto T, Wakabayashi S, Takaoka K (2000) Phosphodiesterase inhibitors, pentoxifylline and rolipram, increase bone mass mainly by promoting bone formation in normal mice. Bone 27:811–817
Takami M, Cho ES, Lee SY, Kamijo R, Yim M (2005) Phosphodiesterase inhibitors stimulate osteoclast formation via TRANCE/RANKL expression in osteoblasts: possible involvement of ERK and p38 MAPK pathways. FEBS Lett 579:832–838
Hausdorff WP, Lohse MJ, Bouvier M, Liggett SB, Caron MG, Lefkowitz RJ (1990) Two kinases mediate agonist-dependent phosphorylation and desensitization of the beta 2-adrenergic receptor. Symp Soc Exp Biol 44:225–240
Lin FT, Daaka Y, Lefkowitz RJ (1998) Beta-arrestins regulate mitogenic signaling and clathrin-mediated endocytosis of the insulin-like growth factor I receptor. J Biol Chem 273:31640–31643
Bliziotes M, Gunness M, Zhang X, Nissenson R, Wiren K (2000) Reduced G-protein-coupled-receptor kinase 2 activity results in impairment of osteoblast function. Bone 27:367–373
Bliziotes M, Murtagh J, Wiren K (1996) Beta-adrenergic receptor kinase-like activity and beta-arrestin are expressed in osteoblastic cells. J Bone Miner Res 11:820–826
Spurney RF, Flannery PJ, Garner SC, Athirakul K, Liu S, Guilak F, Quarles LD (2002) Anabolic effects of a G protein-coupled receptor kinase inhibitor expressed in osteoblasts. J Clin Investig 109:1361–1371
Wang L, Liu S, Quarles LD, Spurney RF (2005) Targeted overexpression of G protein-coupled receptor kinase-2 in osteoblasts promotes bone loss. Am J Physiol Endocrinol Metab 288:E826–E834
Bonnet N, Benhamou CL, Brunet-Imbault B, Arlettaz A, Horcajada MN, Richard O, Vico L, Collomp K, Courteix D (2005) Severe bone alterations under beta2 agonist treatments: bone mass, microarchitecture and strength analyses in female rats. Bone 37:622–633
Bonnet N, Benhamou CL, Beaupied H, Laroche N, Vico L, Dolleans E, Courteix D (2007) Doping dose of salbutamol and exercise: deleterious effect on cancellous and cortical bones in adult rats. J Appl Physiol 102:1502–1509
Kondo A, Mogi M, Koshihara Y, Togari A (2001) Signal transduction system for interleukin-6 and interleukin-11 synthesis stimulated by epinephrine in human osteoblasts and human osteogenic sarcoma cells. Biochem Pharmacol 61:319–326
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17:1231–1234
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA (2011) Matrix-embedded cells control osteoclast formation. Nat Med 17:1235–1241
Fu L, Patel MS, Bradley A, Wagner EF, Karsenty G (2005) The molecular clock mediates leptin-regulated bone formation. Cell 122:803–815
Bonnet N, Benhamou CL, Malaval L, Goncalves C, Vico L, Eder V, Pichon C, Courteix D (2008) Low dose beta-blocker prevents ovariectomy-induced bone loss in rats without affecting heart functions. J Cell Physiol 217:819–827
Kajimura D, Hinoi E, Ferron M, Kode A, Riley KJ, Zhou B, Guo XE, Karsenty G (2011) Genetic determination of the cellular basis of the sympathetic regulation of bone mass accrual. J Exp Med 208:841–851
Bonnet N, Pierroz DD, Ferrari SL (2008) Adrenergic control of bone remodeling and its implications for the treatment of osteoporosis. J Musculoskelet Neuron Interact 8:94–104
Pierroz DD, Bonnet N, Bianchi EN, Bouxsein ML, Baldock PA, Rizzoli R, Ferrari SL (2012) Deletion of beta-adrenergic receptor 1, 2, or both leads to different bone phenotypes and response to mechanical stimulation. J Bone Miner Res 27:1252–1262
Swift JM, Hogan HA, Bloomfield SA (2013) Beta-1 adrenergic agonist mitigates unloading-induced bone loss by maintaining formation. Med Sci Sports Exerc. doi:10.1249/MSS.0b013e31828d39bc
Bouxsein ML, Devlin MJ, Glatt V, Dhillon H, Pierroz DD, Ferrari SL (2009) Mice lacking beta-adrenergic receptors have increased bone mass but are not protected from deleterious skeletal effects of ovariectomy. Endocrinology 150:144–152
Motyl KJ, Bishop KA, Demambro VE, Bornstein SA, Le P, Kawai M, Lotinun S, Horowitz MC, Baron R, Bouxsein ML, Rosen CJ (2013) Altered thermogenesis and impaired bone remodeling in Misty mice. J Bone Miner Res. doi:10.1002/jbmr.1943
Bonnet N, Laroche N, Vico L, Dolleans E, Benhamou CL, Courteix D (2006) Dose effects of propranolol on cancellous and cortical bone in ovariectomized adult rats. J Pharmacol Exp Ther 318:1118–1127
Kondo H, Nifuji A, Takeda S, Ezura Y, Rittling SR, Denhardt DT, Nakashima K, Karsenty G, Noda M (2005) Unloading induces osteoblastic cell suppression and osteoclastic cell activation to lead to bone loss via sympathetic nervous system. J Biol Chem 280:30192–30200
Sato T, Arai M, Goto S, Togari A (2010) Effects of propranolol on bone metabolism in spontaneously hypertensive rats. J Pharmacol Exp Ther 334:99–105
Wang TM, Hsu JF, Jee WS, Matthews JL (1993) Evidence for reduced cancellous bone mass in the spontaneously hypertensive rat. Bone Miner 20:251–264
Gotoh M, Mizuno K, Ono Y, Takahashi M (2005) High blood pressure, bone-mineral loss and insulin resistance in women. Hypertens Res 28:565–570
Young E, Korszun A (2009) Stress, the HPA axis and depressive illness. In: Larry RS (ed) Encyclopedia of neuroscience. Academic Press, Oxford, pp 543–548
Lindenfeld J, Crawford MH, O’Rourke RA, Levine SP, Montiel MM, Horwitz LD (1980) Adrenergic responsiveness after abrupt propranolol withdrawal in normal subjects and in patients with angina pectoris. Circulation 62:704–711
Boudoulas H, Lewis RP, Kates RE, Dalamangas G (1977) Hypersensitivity to adrenergic stimulation after propranolol withdrawal in normal subjects. Ann Intern Med 87:433–436
Granata AR (1985) Prolonged treatment with (+/−) propranolol induces supersensitivity to (l)noradrenaline in mesenteric arteries in the rat. Gen Pharmacol 16:463–468
Karliner JS (1989) Effects of beta-blockade on beta-adrenergic receptors and signal transduction. J Cardiovasc Pharmacol 14(Suppl 5):S6–S12
Tse J, Wrenn RW, Kuo JF (1980) Thyroxine-induced changes in characteristics and activities of beta-adrenergic receptors and adenosine 3′,5′-monophosphate and guanosine 3′,5′-monophosphate systems in the heart may be related to reputed catecholamine supersensitivity in hyperthyroidism. Endocrinology 107:6–16
Aarons RD, Nies AS, Gal J, Hegstrand LR, Molinoff PB (1980) Elevation of beta-adrenergic receptor density in human lymphocytes after propranolol administration. J Clin Investig 65:949–957
Ma Y, Nyman JS, Tao H, Moss HH, Yang X, Elefteriou F (2011) Beta2-adrenergic receptor signaling in osteoblasts contributes to the catabolic effect of glucocorticoids on bone. Endocrinology 152:1412–1422
Robbins J, Hirsch C, Whitmer R, Cauley J, Harris T (2001) The association of bone mineral density and depression in an older population. J Am Geriatr Soc 49:732–736
Cizza G, Primma S, Csako G (2009) Depression as a risk factor for osteoporosis. Trends Endocrinol Metab 20:367–373
Michelson D, Stratakis C, Hill L, Reynolds J, Galliven E, Chrousos G, Gold P (1996) Bone mineral density in women with depression. N Engl J Med 335:1176–1181
Yazici KM, Akinci A, Sutcu A, Ozcakar L (2003) Bone mineral density in premenopausal women with major depressive disorder. Psychiatry Res 117:271–275
Kahl KG, Rudolf S, Stoeckelhuber BM, Dibbelt L, Gehl HB, Markhof K, Hohagen F, Schweiger U (2005) Bone mineral density, markers of bone turnover, and cytokines in young women with borderline personality disorder with and without comorbid major depressive disorder. Am J Psychiatry 162:168–174
Ensrud KE, Blackwell T, Mangione CM, Bowman PJ, Bauer DC, Schwartz A, Hanlon JT, Nevitt MC, Whooley MA, Study of Osteoporotic Fractures Research G (2003) Central nervous system active medications and risk for fractures in older women. Arch Intern Med 163:949–957
Esler M, Rumantir M, Wiesner G, Kaye D, Hastings J, Lambert G (2001) Sympathetic nervous system and insulin resistance: from obesity to diabetes. Am J Hypertens 14:304S–309S
Lutgendorf SK, DeGeest K, Dahmoush L, Farley D, Penedo F, Bender D, Goodheart M, Buekers TE, Mendez L, Krueger G, Clevenger L, Lubaroff DM, Sood AK, Cole SW (2011) Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav Immun 25:250–255
Hughes JW, Watkins L, Blumenthal JA, Kuhn C, Sherwood A (2004) Depression and anxiety symptoms are related to increased 24-hour urinary norepinephrine excretion among healthy middle-aged women. J Psychosom Res 57:353–358
Yirmiya R, Goshen I, Bajayo A, Kreisel T, Feldman S, Tam J, Trembovler V, Csernus V, Shohami E, Bab I (2006) Depression induces bone loss through stimulation of the sympathetic nervous system. Proc Natl Acad Sci USA 103:16876–16881
Campbell JP, Karolak MR, Ma Y, Perrien DS, Masood-Campbell SK, Penner NL, Munoz SA, Zijlstra A, Yang X, Sterling JA, Elefteriou F (2012) Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol 10:e1001363
Powe DG, Voss MJ, Zanker KS, Habashy HO, Green AR, Ellis IO, Entschladen F (2010) Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 1:628–638
Ganz PA, Habel LA, Weltzien EK, Caan BJ, Cole SW (2011) Examining the influence of beta blockers and ACE inhibitors on the risk for breast cancer recurrence: results from the LACE cohort. Breast Cancer Res Treat 129:549–556
Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD, Hortobagyi GN, Gonzalez-Angulo AM (2011) Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol 29:2645–2652
Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K (2011) Beta blockers and breast cancer mortality: a population-based study. J Clin Oncol 29:2635–2644
Hinoi E, Gao N, Jung DY, Yadav V, Yoshizawa T, Myers MG Jr, Chua SC Jr, Kim JK, Kaestner KH, Karsenty G (2008) The sympathetic tone mediates leptin’s inhibition of insulin secretion by modulating osteocalcin bioactivity. J Cell Biol 183:1235–1242
Altman JD, Trendelenburg AU, MacMillan L, Bernstein D, Limbird L, Starke K, Kobilka BK, Hein L (1999) Abnormal regulation of the sympathetic nervous system in alpha2A-adrenergic receptor knockout mice. Mol Pharmacol 56:154–161
Han J, Zou Z, Zhu C, Deng J, Wang J, Ran X, Shi C, Ai G, Li R, Cheng T, Su Y (2009) DNA synthesis of rat bone marrow mesenchymal stem cells through alpha1-adrenergic receptors. Arch Biochem Biophys 490:96–102
Nishiura T, Abe K (2007) Alpha1-adrenergic receptor stimulation induces the expression of receptor activator of nuclear factor kappaB ligand gene via protein kinase C and extracellular signal-regulated kinase pathways in MC3T3-E1 osteoblast-like cells. Arch Oral Biol 52:778–785
Huang HH, Brennan TC, Muir MM, Mason RS (2009) Functional alpha1- and beta2-adrenergic receptors in human osteoblasts. J Cell Physiol 220:267–275
Choi YJ, Lee JY, Lee SJ, Chung CP, Park YJ (2011) Alpha-adrenergic blocker mediated osteoblastic stem cell differentiation. Biochem Biophys Res Commun 416:232–238
Idris AI, van ‘t Hof RJ, Greig IR, Ridge SA, Baker D, Ross RA, Ralston SH (2005) Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat Med 11:774–779
Tam J, Ofek O, Fride E, Ledent C, Gabet Y, Muller R, Zimmer A, Mackie K, Mechoulam R, Shohami E, Bab I (2006) Involvement of neuronal cannabinoid receptor CB1 in regulation of bone mass and bone remodeling. Mol Pharmacol 70:786–792
Wong PK, Christie JJ, Wark JD (2007) The effects of smoking on bone health. Clin Sci 113:233–241
Walker LM, Preston MR, Magnay JL, Thomas PB, El Haj AJ (2001) Nicotinic regulation of c-fos and osteopontin expression in human-derived osteoblast-like cells and human trabecular bone organ culture. Bone 28:603–608
Rothem DE, Rothem L, Soudry M, Dahan A, Eliakim R (2009) Nicotine modulates bone metabolism-associated gene expression in osteoblast cells. J Bone Miner Metab 27:555–561
Bajayo A, Bar A, Denes A, Bachar M, Kram V, Attar-Namdar M, Zallone A, Kovacs KJ, Yirmiya R, Bab I (2012) Skeletal parasympathetic innervation communicates central IL-1 signals regulating bone mass accrual. Proc Natl Acad Sci USA 109:15455–15460
Shi Y, Oury F, Yadav VK, Wess J, Liu XS, Guo XE, Murshed M, Karsenty G (2010) Signaling through the M(3) muscarinic receptor favors bone mass accrual by decreasing sympathetic activity. Cell Metab 11:231–238
Reid IR, Gamble GD, Grey AB, Black DM, Ensrud KE, Browner WS, Bauer DC (2005) Beta-blocker use, BMD, and fractures in the study of osteoporotic fractures. J Bone Miner Res 20:613–618
Rejnmark L, Vestergaard P, Kassem M, Christoffersen BR, Kolthoff N, Brixen K, Mosekilde L (2004) Fracture risk in perimenopausal women treated with beta-blockers. Calcif Tissue Int 75:365–372
Graham S, Hammond-Jones D, Gamie Z, Polyzois I, Tsiridis E, Tsiridis E (2008) The effect of beta-blockers on bone metabolism as potential drugs under investigation for osteoporosis and fracture healing. Expert Opin Investig Drugs 17:1281–1299
Reid IR (2008) Effects of beta-blockers on fracture risk. J Musculoskelet Neuron Interact 8:105–110
Bonnet N, Gadois C, McCloskey E, Lemineur G, Lespessailles E, Courteix D, Benhamou CL (2007) Protective effect of beta blockers in postmenopausal women: influence on fractures, bone density, micro and macroarchitecture. Bone 40:1209–1216
Reid IR, Lucas J, Wattie D, Horne A, Bolland M, Gamble GD, Davidson JS, Grey AB (2005) Effects of a beta-blocker on bone turnover in normal postmenopausal women: a randomized controlled trial. J Clin Endocrinol Metab 90:5212–5216
Levasseur R, Legrand E, Chappard D, Audran M (2005) Central control of bone mass: potential therapeutic implications. Jt Bone Spine 72:474–476
Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G (2003) Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology 60:1327–1332
Robertson D, Haile V, Perry SE, Robertson RM, Phillips JA III, Biaggioni I (1991) Dopamine beta-hydroxylase deficiency. A genetic disorder of cardiovascular regulation. Hypertension 18:1–8
Biaggioni I, Robertson D (1987) Endogenous restoration of noradrenaline by precursor therapy in dopamine-beta-hydroxylase deficiency. Lancet 2:1170–1172
Man in ‘t Veld AJ, Boomsma F, Moleman P, Schalekamp MA (1987) Congenital dopamine-beta-hydroxylase deficiency. A novel orthostatic syndrome. Lancet 1:183–188
Tinetti ME, Speechley M, Ginter SF (1988) Risk factors for falls among elderly persons living in the community. N Engl J Med 319:1701–1707
Graafmans WC, Ooms ME, Hofstee HM, Bezemer PD, Bouter LM, Lips P (1996) Falls in the elderly: a prospective study of risk factors and risk profiles. Am J Epidemiol 143:1129–1136
Robertson D (1999) The epidemic of orthostatic tachycardia and orthostatic intolerance. Am J Med Sci 317:75–77
Low PA, Opfer-Gehrking TL, Textor SC, Benarroch EE, Shen WK, Schondorf R, Suarez GA, Rummans TA (1995) Postural tachycardia syndrome (POTS). Neurology 45:S19–S25
Benarroch EE (2012) Postural tachycardia syndrome: a heterogeneous and multifactorial disorder. Mayo Clin Proc 87:1214–1225
de Vries F, Pouwels S, Bracke M, Leufkens HG, Cooper C, Lammers JW, van Staa TP (2007) Use of beta-2 agonists and risk of hip/femur fracture: a population-based case-control study. Pharmacoepidemiol Drug Saf 16:612–619
Veldhuis-Vlug AG, El Mahdiui M, Endert E, Heijboer AC, Fliers E, Bisschop PH (2012) Bone resorption is increased in pheochromocytoma patients and normalizes following adrenalectomy. J Clin Endocrinol Metab 97:E2093–E2097
Kado DM, Lui LY, Cummings SR, Study of Osteoporotic Fractures Research Group (2002) Rapid resting heart rate: a simple and powerful predictor of osteoporotic fractures and mortality in older women. J Am Geriatr Soc 50:455–460
Tosun A, Dogru MT, Aydn G, Keles I, Arslan A, Guneri M, Orkun S, Ebinc H (2011) Does autonomic dysfunction exist in postmenopausal osteoporosis? Am J Phys Med Rehabil 90:1012–1019
Farr JN, Charkoudian N, Barnes JN, Monroe DG, McCready LK, Atkinson EJ, Amin S, Melton LJ III, Joyner MJ, Khosla S (2012) Relationship of sympathetic activity to bone microstructure, turnover, and plasma osteopontin levels in women. J Clin Endocrinol Metab 97:4219–4227
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors report that they have no conflict of interest.
Rights and permissions
About this article
Cite this article
Elefteriou, F., Campbell, P. & Ma, Y. Control of Bone Remodeling by the Peripheral Sympathetic Nervous System. Calcif Tissue Int 94, 140–151 (2014). https://doi.org/10.1007/s00223-013-9752-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-013-9752-4