
Abstract
Vascular calcification is highly associated with cardiovascular disease mortality, particularly in high-risk patients with diabetes and chronic kidney diseases (CKD). In blood vessels, intimal calcification is associated with atherosclerosis, whereas medial calcification is a nonocclusive process which leads to increased vascular stiffness and reduced vascular compliance. In the valves, calcification of the leaflets can change the mechanical properties of the tissue and result in stenosis. For many decades, vascular calcification has been noted as a consequence of aging. Studies now confirm that vascular calcification is an actively regulated process and shares many features with bone development and metabolism. This review provides an update on the mechanisms of vascular calcification including the emerging roles of the RANK/RANKL/OPG triad, osteoclasts, and microRNAs. Potential treatments adapted from osteoporosis and CKD treatments that are under investigation for preventing and/or regressing vascular calcification are also reviewed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Vascular Calcification
Vascular calcification is the pathological deposition of mineral in the vascular system. It has a variety of forms, including intimal calcification and medial calcification, but can also be found in the valves of the heart. Vascular calcification is associated with atherosclerosis, diabetes, certain hereditary conditions, and kidney disease, especially chronic kidney disease (CKD). Patients with vascular calcification are at higher risk for adverse cardiovascular events. Vascular calcification affects a wide variety of patients. Idiopathic infantile arterial calcification is a rare form of vascular calcification where the arteries of neonates calcify. It is associated with certain genetic mutations and often results in death. In patients with renal failure undergoing hemodialysis, vascular calcification is a frequent complication, with time on hemodialysis associated with more severe calcification. Calcific uremic arteriolopathy or calciphylaxis is another complication of hemodialysis, where arterioles calcify, leading to necrosis of the skin and ulceration, and has a mortality rate of over 50 % at 2 years [reviewed in 1].
Intimal Calcification vs. Medial Calcification
Calcification of the vessel wall occurs in the intimal and medial layers. The intimal layer of the vessel wall is normally composed of endothelial cells and a small amount of subendothelial connective tissue. In atherosclerosis, the intima becomes greatly inflamed and thickened and calcification occurs. Calcification of coronary arteries is an excellent predictor of atherosclerotic plaque burden and may contribute to atherosclerotic plaque rupture, though the connection between atherosclerotic plaque rupture and calcification is heavily debated. Several studies show a link between high coronary artery calcium and risk of cardiac events and mortality, yet some studies have suggested that the most calcified plaques may be more stable and that the plaques most vulnerable to rupture may be those which have a mixed composition of calcified and uncalcified tissue [2].
The medial layer of the vessel wall is composed of smooth muscle cells and elastin-rich extracellular matrix. Calcification of the media occurs preferentially along the elastic lamina, as opposed to the diffuse localization seen in intimal calcification, and is associated with diabetes, kidney disease, hypertension, and osteoporosis. The result of medial calcification is a stiffening of the artery wall, with the associated rise in blood pressure, and a higher risk of cardiovascular mortality [1].
Valves: Native and Bioprosthetic Valves
Vascular calcification can also lead to dysfunction of the heart valves, both native valves and bioprosthetic valves. In the valves, calcification of the leaflets can change the mechanical properties of the tissue and result in stenosis. Calcification of the valves is commonly associated with aging and is the most common pathology seen in excised native valves [3]. Diseased native valves are often treated by valve replacement surgery, with either mechanical or bioprosthetic valves. Bioprosthetic valves have excellent hemodynamic properties and do not require long-term administration of anticoagulants. However, bioprosthetic valves are prone to calcification and may require replacement after 5–10 years. In fact, calcification and its contribution to tissue deterioration is the major mode of failure of bioprosthetic valves. Calcification of both native and bioprosthetic valves impairs cardiac function due to the valves’ inability to properly regulate flow. Valve calcification is also linked to increased risk of cardiac events and mortality even if the patient is asymptomatic [4].
Mechanisms of Vascular Calcification
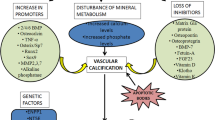
Vascular calcification has been noted as a consequence of aging for many decades. Growing evidence now suggests that vascular calcification, similar to bone remodeling, is an actively regulated process, including both inductive and inhibitory processes [5]. Bone-related proteins, such as alkaline phosphatase, osteocalcin, osteopontin, Runx2, and matrix vesicles, which nucleate hydroxyapatite mineral crystals, have all been observed in calcified valvular lesions. In addition, outright cartilage and bone formation have been identified [6, 7]. The identification of genes that cause ectopic calcification disorders in humans and/or mice has contributed to our knowledge about the regulation of vascular calcification. Remarkably, single-gene mutations in the type II TGFβ receptor and Notch 1 are associated with bicuspid aortic valve and valve disease [8]. Additionally, in vivo and in vitro models have been developed to mimic important aspects of vascular and valvular calcification and have identified new pathways important for this process. The current major mechanisms of vascular calcification based on these studies are summarized in Fig. 1, including (1) failed anticalcific processes, (2) induction of osteochondrogenesis, (3) cell death, (4) abnormal Ca/Pi homeostasis, (5) circulating calciprotein particles, and (6) matrix degradation/modification. The studies supporting these mechanisms have been reviewed in detail in recent articles [9–12]. In this article, we focus on recent evidence for new mechanisms that regulate vascular calcification, including the potential role of the RANK/RANKL/OPG axis, osteoclasts, and microRNAs (miRNA). Finally, we review potential treatments that are under investigation for preventing and/or regressing vascular calcification.

Major mechanisms and mediators of vascular calcification
Potential Role of the RANK/RANKL/OPG Axis and Osteoclasts
Most of the regulatory factors identified to date are thought to contribute to ectopic calcification by either promoting or inhibiting crystal nucleation or growth, cell death, or osteogenic differentiation of mesenchymal cells, rather than affecting resorption, or removal, of the mineral after it has been formed. Osteoclasts are multinucleated, bone resorptive cells derived from bone marrow that play an important role in skeletal mass regulation. In bones, overactive osteoclastic activity leads to bone loss (osteoporosis), and osteoclast deficiency leads to bone overgrowth (osteopetrosis). RANK is a type I membrane protein expressed on the surface of osteoclasts and is involved in their activation upon ligand (RANKL) binding. Osteoprotegerin (OPG) produced by osteoblasts is a potent inhibitor of osteoclast differentiation and survival by acting as a decoy receptor for RANKL, leading to decreased signaling through its membrane receptor, RANK [13].
Growing evidence suggests that the triad of RANK/RANKL/OPG, key proteins involved in bone metabolism, may be important in vascular calcification. OPG, RANKL, and RANK are present in atherosclerotic plaques and valve disease; and their relative expression levels are different depending on the stage of the disease [14, 15]. OPG appears to be protective against vascular calcification since OPG–/– mice developed spontaneous arterial calcification [16] and depleting OPG in ApoE–/– mice increased atherosclerotic lesion progression and calcification [17]. Likewise, evidence that RANKL stimulates vascular calcification is growing. In one study, RANKL increased vascular smooth muscle cell (VSMC) calcification directly by binding to RANK and increasing BMP4 production through the alternative NF-κB pathway [18]. In another study, RANKL indirectly promoted smooth muscle cell calcification by enhancing macrophage paracrine procalcific activity through release of Il-6 and TNFa [19]. These studies suggest that RANK/RANKL may be important in promoting vascular calcification, while OPG inhibits vascular calcification.
Whether or not osteoclasts or osteoclast deficiency might play a role in vascular calcification is less clear. Osteoclast-like cells that express tartrate-resistant acid phosphatase (TRAP) have been found in calcified vascular lesions in humans but were typically present at very low levels and only at advanced stages of disease [6, 20]. Since osteoclasts typically function to resorb bone, a deficiency of osteoclast-like cells as observed in human calcified lesions might be expected to facilitate vascular calcium accrual. However, in ApoE–/– mice, osteoclast-like cells were observed in atherosclerotic lesions, and selective knockdown of Runx2 in smooth muscle cells led to decreased RANKL expression, osteoclast-like cell number, and lesion calcification, suggesting that osteoclasts might actually promote vascular calcification [21]. Clearly, further studies to determine the role of osteoclasts in vascular calcification are needed to distinguish between these possibilities.
MicroRNAs: New Players in Vascular Calcification
miRNA are noncoding, single-stranded RNAs of ~22 nucleotides that are recognized as a novel class of gene regulators. miRNAs bind to complementary sequences on target mRNAs, usually resulting in translational repression or target degradation. One single miRNA may target multiple genes, providing extensive translational regulation; and multiple miRNAs may work together to promote combinatorial regulation by individually targeting many components of a pathway. The role of miRNAs in cardiovascular biology is currently under intense investigation, and specific miRNAs have been associated with various cardiovascular disorders, including vascular remodeling, cardiac hypertrophy, heart failure, and post–myocardial infarction remodeling [22]. Increasing evidence shows that miRNAs may play a pivotal role in regulating vascular cell functions and contribute to vascular calcification. For example, miR-125b, previously found to be involved in osteoblast differentiation, was found to be involved in the osteogenic transdifferentiation of VSMCs by targeting SP7 in vivo and in vitro [23]. miR-204 was identified as an important regulator of VSMC functions by downregulating its target gene, Runx2, and inhibiting medial artery calcification in vivo [24]. Using the kl/kl mouse model and specific miRNA inhibitors, miR-135a*, miR-762, miR-714, and miR-712* were shown to target Ca2+ transporters that are involved in VSMC calcification [25]. These studies implicate miRNAs as a novel link in the mechanisms of vascular calcification.
Potential Treatments of Vascular Calcification
To date, a variety of therapies have been tested to try and prevent or regress vascular calcification. Preventative therapies must be given to at-risk populations prior to the formation or progression of vascular calcification, whereas a therapy capable of eliminating or reducing vascular calcification has the advantage of treating patients who have already developed it. The available therapies are often existing treatments for related conditions and can be categorized as osteoporosis therapies, CKD therapies, and cardiovascular disease therapies. There are also experimental therapies at various research stages. These therapies are outlined in Table 1, which describes the therapies and their mechanism of action as well as any evidence of efficacy.
Osteoporosis Therapies
Bisphosphonates (Pyrophosphate Analogs)
Bisphosphonates have been used as an effective therapy for osteoporosis for almost 40 years. They inhibit bone resorption by binding to hydroxyapatite, where they are ingested by osteoclasts. The uptake of these pyrophosphate analogs interferes with normal osteoclast functions and reduces bone resorption. Pyrophosphate analogs are also extremely potent inhibitors of hydroxyapatite nucleation and crystal growth [26]. Due to the high association of osteoporosis and vascular calcification, bisphosphonates were viewed as a potential therapy for vascular calcification. Pyrophosphate and bisphosphonates have been found to inhibit both vascular calcification and calciphylaxis in several animal studies [27]. It has been hypothesized that this effect is due to lowered serum levels of calcium and phosphate and/or reduced nucleation and growth of calcium phosphate crystals that lead to vascular calcification. Bisphosphonates may be useful for treating calciphylaxis, with a small prospective study suggesting that intravenous ibandronate halted progression and improved healing in eight patients with calciphylaxis [28]. For vascular calcification, even though bisphosphonate therapy has shown effectiveness in animal models, there is still some debate over its efficacy in humans, and the results differ by the specific bisphosphonate in question. In studies of patients with end-stage renal disease undergoing dialysis, etidronate has been found to limit the further progression of vascular calcification [29]. On the other hand, alendronate and ibandronate have had mixed results at limiting the progression of vascular calcification, with animal studies suggesting that they limit vascular calcification, while randomized controlled trials found no effect in humans [30]. The pros of bisphosphonate therapy need to be weighed with the cons, and bisphosphonates are not advisable for CKD patients due to side effects.
Denosumab
Denosumab is a human monoclonal antibody targeting RANKL. RANKL is required for osteoclast differentiation and survival, so denosumab inhibits bone resorption and is one of the newest therapies for osteoporosis. As described above, RANKL also has direct effects on promoting VSMC calcification [18] and TRAP+ osteoclast-like cell formation [21]. Because of these activities, denosumab has also been studied for its ability to prevent vascular calcification. In a mouse model of glucocorticoid-induced osteoporosis, denosumab reduced the amount of calcium deposited in the aorta [31]. Studies in humans are needed, to determine if denosumab is able to prevent vascular calcification.
Osteoprotegerin
OPG is a protein that inhibits bone resorption. It is a decoy receptor that binds and inhibits RANKL and, like denosumab, prevents osteoclastogenesis. OPG is likely involved in vascular calcification, and it inhibits calcification in vitro; it is thought that this activity comes from OPG increasing expression of insulin-like growth factor-I receptor on smooth muscle cells [32]. One study found that OPG knockout mice developed extensive vascular calcification in addition to the expected result of osteoporosis [16]. Treatment with a recombinant fusion protein, Fc-OPG, has been shown to inhibit vascular calcification in animal studies [33].
Teriparatide (Human Parathyroid Hormone 1–34)
Teriparatide is a recombinant protein representing a portion of human parathyroid hormone (PTH), specifically amino acids 1–34. It has been used clinically to increase bone mineral density in patients with osteoporosis, with some studies showing greater efficacy than bisphosphonates [34]. Due to the role of PTH in regulating calcium and phosphate levels, it has been studied as a method to prevent vascular calcification. One study in diabetic low-density lipoprotein receptor–deficient mice found that daily subcutaneous administration of teriparatide greatly reduced the extent of both aortic and cardiac valve calcification [35]. It was also found to increase expression of osteopontin, a potent circulating inhibitor of calcification.
CKD Therapies
Phosphate Binders
Phosphate binders are a class of drugs that bind phosphate and form an insoluble complex in the digestive system. This prevents the phosphate from ever being absorbed, which is beneficial for patients with end-stage renal disease on dialysis whose kidneys are unable to maintain proper phosphate homeostasis [36]. There are two main types of phosphate binders: calcium-containing binders, such as calcium acetate, and calcium-free binders, such as lanthanum carbonate and sevelamer carbonate. Although both types are effective at regulating phosphate levels, they differ in their effect on vascular calcification, with the calcium-containing phosphate binders being thought to contribute to the development and progression of vascular calcification and the calcium-free phosphate binders being thought to slow progression. This hypothesis is supported by several studies in human CKD patients [37]. Most patients on dialysis are currently taking some form of phosphate binder, but the data suggest that non-calcium-containing phosphate binders should be used in patients with high risk for vascular calcification.
Vitamin D Receptor Agonists
Vitamin D plays a role in multiple metabolic pathways, including regulation of mineral metabolism. Vitamin D receptor agonists (VDRAs) are given to patients with CKD to ameliorate the effects of hyperparathyroidism and vitamin D deficiency. Vitamin D deficiency is associated with early-mortality CKD patients, and VDRA therapy prolongs survival in hemodialysis patients; but their mechanism is not fully understood [38]. In a mouse model of CKD, VDRA therapy significantly reduced aortic calcification [39]. In this study, the effect of VDRA therapy was associated with increased expression of serum klotho, a phosphaturic hormone, and vascular medial osteopontin, an inhibitor of vascular calcification.
Calcimimetics
Calcimimetics are compounds that mimic the effects of calcium in vivo. They have been used to treat hyperparathyroidism by binding to calcium-sensing receptors (CaRs) in the parathyroid glands. Calcimimetics are allosteric modulators of CaRs, which make the CaRs more sensitive to existing levels of circulating calcium, which in turn suppresses secretion of PTH. Calcimimetics have been shown to reduce vascular calcification in a variety of uremic rat models [40]. There is also evidence of efficacy in humans. The ADVANCE study found that calcimimetics with VDRA therapy slowed the progression of vascular calcification over VDRA therapy alone in hemodialysis [41]. Calcimimetics may be an important tool to treat vascular calcification, but it is unclear if they will work in non-CKD patients.
Vitamin K
Vitamin K is a group of cofactor vitamins involved in coagulation and various metabolic pathways. It is required by the enzyme gamma-glutamate carboxylase to modify matrix Gla protein (MGP) into its active carboxylated form [42]. Active MGP inhibits calcification, and the exact mechanism is still being elucidated. Possibilities include binding to crystals and halting further growth or binding to bone morphogenic protein and preventing osteogenic differentiation of cells. Vitamin K was found to inhibit vascular calcification in warfarin-treated rats [43]. In humans, increased expression of undercarboxylated MGP is associated with vascular calcification and increased risk of cardiovascular mortality, and supplementation with vitamin K2 was found to increase levels of carboxylated MGP in dialysis patients [44]. Further study is necessary to determine if the increased levels of active MGP will lead to prevention of vascular calcification in humans.
Sodium Thiosulfate
Sodium thiosulfate is a small molecule that acts as a vasodilator, antioxidant, and calcium chelator in vivo. It has been used successfully to treat calciphylaxis, where it reduces calcification and improves local circulation. It binds to calcium to form calcium thiosulfate, which can be excreted from the body [45]. Due to its success in treating calciphylaxis, it has been studied for treatment of other forms of vascular calcification. In a uremic rat model of vascular calcification, treatment with sodium thiosulfate completely prevented aortic calcification; however, this was associated with lowered bone strength in the treated animals [46]. One pilot study of sodium thiosulfate treatment in patients undergoing hemodialysis suggested that it may be safe to use, with few side effects seen after 5 months of treatment; larger studies are needed to confirm efficacy [47].
Cardiovascular Disease Therapies
Endothelin Receptor Antagonists
The endothelin receptor is involved with regulating blood pressure; when endothelins bind to this receptor blood pressure increases due to vasoconstriction. For this reason, endothelin receptor antagonists (ERAs), which block the binding of endothelins, are popular therapies for hypertension. The receptor is also important in vascular calcification, with one study showing an increase in levels of endothelin in vitro and in vivo; this calcification could be ameliorated by ERAs [48]. Studies in rat models of hypertension found that darusentan reduced aortic calcification [49]. Studies in humans are needed before the effect of ERA therapy on vascular calcification can be ascertained, but this method shows promise.
Statins
Statins are a group of drugs that help to lower serum cholesterol by inhibiting HMG-CoA reductase in the liver, to help prevent the formation of atherosclerotic lesions. Valve stenosis and calcification have some similarities to atherosclerosis, including inflammation and lipid deposition; and both atherosclerosis and valve calcification often appear in the same patients [50]. For these reasons, statin therapy was investigated as a potential treatment to prevent calcific valve stenosis. Early retrospective studies suggested a benefit of statins, with treated patients exhibiting reduced progression of calcific aortic stenosis [51]. However, when controlled randomized trials were conducted, there was no benefit seen. It is now generally accepted that statin therapy does not impact calcific aortic stenosis [50].
Experimental Therapies
EDTA Chelation Therapy
Ethylenediaminetetraacetic acid (EDTA) is a synthetic amino acid which is capable of binding free calcium. It has various commercial and scientific applications, but its usefulness in medicine is unclear. Because it can bind calcium, it has been proposed as a therapy for vascular calcification. One small human trial has shown a regression of vascular calcification in some patients as measured by computed tomography [52]. However, this study lacked a control group for comparison; also, fewer than half of the patients responded to the therapy, so further study is needed.
Metabolic Acidosis
Metabolic acidosis is a condition where the pH of the blood and bodily fluids is abnormally low. It is commonly seen in patients with CKD and can result in a plethora of negative side effects including bone disease, muscle degradation, inflammation, and cardiovascular disease. It has been suggested that metabolic acidosis may have a beneficial effect on preventing calcification in the vasculature through the same chemical and cellular mechanisms that result in bone dissolution. Specifically, metabolic acidosis is known to result in the physicochemical dissolution of bone mineral as well as the activation of osteoclasts through signaling pathways such as RANKL [53]. A study of calcitriol-induced calcification in uremic rats found that metabolic acidosis helped to prevent calcification in vitro and in vivo [54]. It is unclear if the beneficial effects of metabolic acidosis can be separated from the negative effects.
Autologous Osteoclasts
Osteoclasts are the main mineral-resorbing cells in the body. They are formed from fusing mononuclear precursor cells of the myeloid lineage and differentiate in response to cytokines including macrophage colony-stimulating factor (M-CSF) and RANKL [55]. A landmark study found that osteoclasts derived from rat bone marrow were capable of removing mineral from calcified elastin in vitro; additionally, when the cells were coimplanted with elastin subdermally in rats, calcification was limited [56]. These studies were the first to suggest that autologous osteoclasts might be utilized as a therapy for vascular calcification. Methods for local delivery and activation of osteoclasts at sites of vascular calcification in vivo would aid in the development of this type of cell therapy. In our lab, we have engineered monocytic precursors to differentiate into osteoclasts under the control of a small molecule. This differentiation is independent of RANKL and M-CSF and resistant to inhibition by OPG. When combined with autologous precursors, this system could be used to develop a local cell-based therapy to treat or prevent ectopic calcification (Wu et al., unpublished data).
Future Challenges
Vascular calcification was first recognized more than a century ago. In the last few decades, advances in the field have improved our understanding of the molecular mechanisms that contribute to vascular calcification. Most recently, several lines of evidence have inversely correlated vascular calcification with bone formation, and several potential treatments under investigation are related to osteoporosis therapies. However, the efficacy of these drugs at inhibiting vascular calcification was discovered from either studies of several animal models or a relatively small set of human trials and may be more based on their efficacy at blocking calcium phosphate crystal growth, rather than their inhibition of osteoclast function. Further studies using larger numbers of patients are needed to reveal the action of these drugs. In addition, therapeutic strategies used in CKD patients to reduce phosphate burden or block calcium channels, while showing promise in preventing the progression of vascular calcification, may have potential risks for bone health. Thus, for the development of new treatments specifically targeting vascular calcification, serious consideration must be given to the implications for bone metabolism, to avoid potentially harmful effects to bone health.
References
Giachelli CM (2004) Mechanisms of vascular calcification in uremia. Semin Nephrol 24:401–402
Nicoll R, Henein MY (2012) Arterial calcification: friend or foe? Int J Cardiol (in press)
Butany J, Collins MJ, Demellawy DE, Nair V, Israel N, Leong SW, Borger MA (2005) Morphological and clinical findings in 247 surgically excised native aortic valves. Can J Cardiol 21:747–755
Otto CM, Burwash IG, Legget ME, Munt BI, Fujioka M, Healy NL, Kraft CD, Miyake-Hull CY, Schwaegler RG (1997) Prospective study of asymptomatic valvular aortic stenosis. Clinical, echocardiographic, and exercise predictors of outcome. Circulation 95:2262–2270
Boström KI, Rajamannan NM, Towler DA (2011) The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ Res 109:564–577
Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS (2001) Bone formation and inflammation in cardiac valves. Circulation 103:1522–1528
Steiner I, Kasparová P, Kohout A, Dominik J (2007) Bone formation in cardiac valves: a histopathological study of 128 cases. Virchows Arch 450:653–657
O’Brien KD (2007) Epidemiology and genetics of calcific aortic valve disease. J Investig Med 55:284–291
Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM (2011) Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 109:697–711
Chen JH, Simmons CA (2011) Cell–matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res 108:1510–1524
Pai AS, Giachelli CM (2010) Matrix remodeling in vascular calcification associated with chronic kidney disease. J Am Soc Nephrol 21:1637–1640
Sage AP, Tintut Y, Demer LL (2010) Regulatory mechanisms in vascular calcification. Nat Rev Cardiol 7:528–536
Liu C, Walter TS, Huang P, Zhang S, Zhu X, Wu Y, Wedderburn LR, Tang P, Owens RJ, Stuart DI, Ren J, Gao B (2010) Structural and functional insights of RANKL–RANK interaction and signaling. J Immunol 184:6910–6919
Heymann MF, Herisson F, Davaine JM, Charrier C, Battaglia S, Passuti N, Lambert G, Gouëffic Y, Heymann D (2012) Role of the OPG/RANK/RANKL triad in calcifications of the atheromatous plaques: comparison between carotid and femoral beds. Cytokine 58:300–306
Steinmetz M, Skowasch D, Wernert N, Welsch U, Preusse CJ, Welz A, Nickenig G, Bauriedel G (2008) Differential profile of the OPG/RANKL/RANK-system in degenerative aortic native and bioprosthetic valves. J Heart Valve Dis 17:187–193
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS (1998) Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12:1260–1268
Bennett BJ, Scatena M, Kirk EA, Rattazzi M, Varon RM, Averill M, Schwartz SM, Giachelli CM, Rosenfeld ME (2006) Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE–/– mice. Arterioscler Thromb Vasc Biol 26:2117–2124
Panizo S, Cardus A, Encinas M, Parisi E, Valcheva P, López-Ongil S, Coll B, Fernandez E, Valdivielso JM (2009) RANKL increases vascular smooth muscle cell calcification through a RANK-BMP4-dependent pathway. Circ Res 104:1041–1048
Deuell KA, Callegari A, Giachelli CM, Rosenfeld ME, Scatena M (2012) RANKL enhances macrophage paracrine pro-calcific activity in high phosphate-treated smooth muscle cells: dependence on IL-6 and TNF-α. J Vasc Res 49:510–521
Oksala N, Levula M, Pelto-Huikko M, Kytömäki L, Soini JT, Salenius J, Kähönen M, Karhunen PJ, Laaksonen R, Parkkila S, Lehtimäki T (2010) Carbonic anhydrases II and XII are up-regulated in osteoclast-like cells in advanced human atherosclerotic plaques—tampere vascular study. Ann Med 42:360–370
Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, Anderson PG, Tintut Y, Demer LL, Wang D, Chen Y (2011) Runx2-upregulated receptor activator of nuclear factor κB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol 31:1387–1396
Small EM, Olson EN (2011) Pervasive roles of microRNAs in cardiovascular biology. Nature 469:336–342
Goettsch C, Rauner M, Pacyna N, Hempel U, Bornstein SR, Hofbauer LC (2011) miR-125b regulates calcification of vascular smooth muscle cells. Am J Pathol 179:1594–1600
Cui RR, Li SJ, Liu LJ, Yi L, Liang QH, Zhu X, Liu GY, Liu Y, Wu SS, Liao XB, Yuan LQ, Mao DA, Liao EY (2012) MicroRNA-204 regulates vascular smooth muscle cell calcification in vitro and in vivo. Cardiovasc Res 96:320–329
Gui T, Zhou G, Sun Y, Shimokado A, Itoh S, Oikawa K, Muragaki Y (2012) MicroRNAs that target Ca2+ transporters are involved in vascular smooth muscle cell calcification. Lab Invest 92:1250–1259
Boskey A (2003) Bone mineral crystal size. Osteoporos Int 14(Suppl 5):S16–S20
Persy V, De Broe M, Ketteler M (2006) Bisphosphonates prevent experimental vascular calcification: treat the bone to cure the vessels? Kidney Int 70:1537–1538
Torregrosa JV, Durán CE, Barros X, Blasco M, Arias M, Cases A, Campistol JM (2012) Successful treatment of calcific uraemic arteriolopathy with bisphosphonates. Nefrologia 32:329–334
Nitta K, Akiba T, Suzuki K, Uchida K, Watanabe R, Majima K, Aoki T, Nihei H (2004) Effects of cyclic intermittent etidronate therapy on coronary artery calcification in patients receiving long-term hemodialysis. Am J Kidney Dis 44:680–688
Toussaint ND, Lau KK, Strauss BJ, Polkinghorne KR, Kerr PG (2010) Effect of alendronate on vascular calcification in CKD stages 3 and 4: a pilot randomized controlled trial. Am J Kidney Dis 56:57–68
Helas S, Goettsch C, Schoppet M, Zeitz U, Hempel U, Morawietz H, Kostenuik PJ, Erben RG, Hofbauer LC (2009) Inhibition of receptor activator of NF-kappaB ligand by denosumab attenuates vascular calcium deposition in mice. Am J Pathol 175:473–478
Di Bartolo BA, Schoppet M, Mattar MZ, Rachner TD, Shanahan CM, Kavurma MM (2011) Calcium and osteoprotegerin regulate IGF1R expression to inhibit vascular calcification. Cardiovasc Res 91:537–545
Morony S, Tintut Y, Zhang Z, Cattley RC, Van G, Dwyer D, Stolina M, Kostenuik PJ, Demer LL (2008) Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in ldlr–/– mice. Circulation 117:411–420
Saag KG, Shane E, Boonen S, Marín F, Donley DW, Taylor KA, Dalsky GP, Marcus R (2007) Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 357:2028–2039
Shao JS, Cheng SL, Charlton-Kachigian N, Loewy AP, Towler DA (2003) Teriparatide (human parathyroid hormone [1–34]) inhibits osteogenic vascular calcification in diabetic low density lipoprotein receptor–deficient mice. J Biol Chem 278:50195–50202
Hutchison AJ, Smith CP, Brenchley PE (2011) Pharmacology, efficacy and safety of oral phosphate binders. Nat Rev Nephrol 7:578–589
Qunibi W, Moustafa M, Muenz LR, He DY, Kessler PD, Diaz-Buxo JA, Budoff M, CARE-2 Investigators (2008) A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 51:952–965
Wolf M, Shah A, Gutierrez O, Ankers E, Monroy M, Tamez H, Steele D, Chang Y, Camargo CA, Tonelli M, Thadhani R (2007) Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int 72:1004–1013
Lau WL, Leaf EM, Hu MC, Takeno MM, Kuro-o M, Moe OW, Giachelli CM (2012) Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int 82:1261–1270
Jung S, Querfeld U, Müller D, Rudolph B, Peters H, Krämer S (2012) Submaximal suppression of parathyroid hormone ameliorates calcitriol-induced aortic calcification and remodeling and myocardial fibrosis in uremic rats. J Hypertens 30:2182–2191
Raggi P, Chertow GM, Torres PU, Csiky B, Naso A, Nossuli K, Moustafa M, Goodman WG, Lopez N, Downey G, Dehmel B, Floege J, Group AS (2011) The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transpl 26:1327–1339
Westenfeld R, Krueger T, Schlieper G, Cranenburg EC, Magdeleyns EJ, Heidenreich S, Holzmann S, Vermeer C, Jahnen-Dechent W, Ketteler M, Floege J, Schurgers LJ (2012) Effect of vitamin K2 supplementation on functional vitamin K deficiency in hemodialysis patients: a randomized trial. Am J Kidney Dis 59:186–195
Spronk HM, Soute BA, Schurgers LJ, Thijssen HH, De Mey JG, Vermeer C (2003) Tissue-specific utilization of menaquinone-4 results in the prevention of arterial calcification in warfarin-treated rats. J Vasc Res 40:531–537
Schlieper G, Westenfeld R, Krüger T, Cranenburg EC, Magdeleyns EJ, Brandenburg VM, Djuric Z, Damjanovic T, Ketteler M, Vermeer C, Dimkovic N, Floege J, Schurgers LJ (2011) Circulating nonphosphorylated carboxylated matrix gla protein predicts survival in ESRD. J Am Soc Nephrol 22:387–395
Ong S, Coulson IH (2012) Diagnosis and treatment of calciphylaxis. Skinmed 10:166–170
Pasch A, Schaffner T, Huynh-Do U, Frey BM, Frey FJ, Farese S (2008) Sodium thiosulfate prevents vascular calcifications in uremic rats. Kidney Int 74:1444–1453
Mathews SJ, de Las Fuentes L, Podaralla P, Cabellon A, Zheng S, Bierhals A, Spence K, Slatopolsky E, Davila-Roman VG, Delmez JA (2011) Effects of sodium thiosulfate on vascular calcification in end-stage renal disease: a pilot study of feasibility, safety and efficacy. Am J Nephrol 33:131–138
Wu SY, Zhang BH, Pan CS, Jiang HF, Pang YZ, Tang CS, Qi YF (2003) Endothelin-1 is a potent regulator in vivo in vascular calcification and in vitro in calcification of vascular smooth muscle cells. Peptides 24:1149–1156
Essalihi R, Ouellette V, Dao HH, McKee MD, Moreau P (2004) Phenotypic modulation of vascular smooth muscle cells during medial arterial calcification: a role for endothelin? J Cardiovasc Pharmacol 44(Suppl 1):S147–S150
Wierzbicki AS, Viljoen A, Chambers JB (2010) Aortic stenosis and lipids: does intervention work? Curr Opin Cardiol 25:379–384
Novaro GM, Tiong IY, Pearce GL, Lauer MS, Sprecher DL, Griffin BP (2001) Effect of hydroxymethylglutaryl coenzyme a reductase inhibitors on the progression of calcific aortic stenosis. Circulation 104:2205–2209
Maniscalco BS, Taylor KA (2004) Calcification in coronary artery disease can be reversed by EDTA-tetracycline long-term chemotherapy. Pathophysiology 11:95–101
Krieger NS, Frick KK, Bushinsky DA (2004) Mechanism of acid-induced bone resorption. Curr Opin Nephrol Hypertens 13:423–436
Mendoza FJ, Lopez I, Montes de Oca A, Perez J, Rodriguez M, Aguilera-Tejero E (2008) Metabolic acidosis inhibits soft tissue calcification in uremic rats. Kidney Int 73:407–414
Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390:175–179
Simpson CL, Lindley S, Eisenberg C, Basalyga DM, Starcher BC, Simionescu DT, Vyavahare NR (2007) Toward cell therapy for vascular calcification: osteoclast-mediated demineralization of calcified elastin. Cardiovasc Pathol 16:29–37
Acknowledgements
Studies in C. M. G.’s lab are supported by NIH grants R01 HL114611, R01 HL62329, and R01 HL081785; Washington State Life Science Discovery grant 2361524; and a grant from Abbott Laboratories. C.R. is supported by the Bioengineering Cardiovascular NIH training grant T32EB001650.
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors have stated that they have no conflict of interest.
Rights and permissions
About this article
Cite this article
Wu, M., Rementer, C. & Giachelli, C.M. Vascular Calcification: An Update on Mechanisms and Challenges in Treatment. Calcif Tissue Int 93, 365–373 (2013). https://doi.org/10.1007/s00223-013-9712-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-013-9712-z