
Abstract
Vitamin D-dependent rickets type II (VDDR-type II) is a rare disorder caused by mutations in the vitamin D receptor (VDR) gene. Here, we describe a patient with VDDR-type II with severe alopecia and rickets. She had hypocalcemia, hypophosphatemia, secondary hyperparathyroidism, and elevated serum alkaline phosphatase and 1,25-dihydroxyvitamin D3. Sequence analysis of the lymphocyte VDR cDNA revealed deletion mutation c.716delA. Sequence analysis of her genomic DNA fragment amplified from exon 6 of the VDR gene incorporating this mutation confirmed the presence of the mutation in homozygous form. This frameshift mutation in the ligand binding domain (LBD) resulted in premature termination (p.Lys240Argfs) of the VDR protein. The mutant protein contained 246 amino acids, with 239 normal amino acids at the N terminus, followed by seven changed amino acids resulting in complete loss of its LBD. The mutant VDR protein showed evidence of 50% reduced binding with VDR response elements on electrophoretic mobility assay in comparison to the wild-type VDR protein. She was treated with high-dose calcium infusion and oral phosphate. After 18 months of treatment, she gained 6 cm of height, serum calcium and phosphorus improved, alkaline phosphatase levels decreased, and intact PTH normalized. Radiologically, there were signs of healing of rickets. Her parents and one of her siblings had the same c.716delA mutation in heterozygous form. Despite the complete absence of LBD, the rickets showed signs of healing with intravenous calcium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Vitamin D-dependent rickets type II (VDDR-type II), also known as hereditary vitamin D-resistant rickets, is a rare autosomal recessive disorder characterized by rickets, alopecia, and resistance to vitamin D therapy [1, 2]. The resistance to 1,25(OH)2D is caused by heterogeneous mutations in the vitamin D receptor (VDR) [1]. VDR is a member of the nuclear receptor family. Baker et al. [3] first described the normal sequence of human VDR cDNA. Its gene is located on chromosome 12q13.11 and spans approximately 75 kb of genomic DNA. It has two mRNA transcript variants, i.e., one (shorter) and two (longer), both of which code for the same VDR protein of 427 amino acids. Variant 1 is a 4,669 bp-long mRNA and has two noncoding untranslated exons at the 5′ region, followed by eight coding exons (NCBI, reference sequence NM_000376.2). Variant 2 mRNA is a 4,791 bp-long transcript with three untranslated exons and eight coding exons (reference sequence NM_001017535.1). The structure of various domains of the VDR protein is similar to that of other nuclear receptors and includes an N-terminal A/B regulatory domain containing the activation function 1 (AF-1) region, which leads to a weak but ligand-independent transcription, DNA binding domain (DBD), hinge domain, and ligand binding domain (LBD) [1].
Hughes et al. [4] first described a case of VDDR-type II linked with a mutation in exon 3 of VDR. Since then, to the best of our knowledge, 40 different mutations in the VDR gene have been described in patients with VDDR-type II [1, 5–24]. Malloy et al. [1] reviewed the first 21 of these mutations reported up to 1999. Subsequently, 19 mutations have been described [5–17]. The majority of these were point mutations and others include deletion/insertion/duplication mutations involving multiple base pairs. Major deletion involving exons 7–9 has also been reported in one case [1]. Three of these were due to compound heterozygous mutations [5–7]. To date, single-nucleotide deletion has been described in only one patient with VDDR-type II as part of a compound heterozygous mutation [6]. There is no hot spot for these point mutations as revealed by the fact that 12 of the above mutations were in the DBD, 21 in the LBD, one in the hinge region [18], and two in the splice site [17, 20].
Most patients with VDDR-type II and VDR mutations have been from the Middle East countries and Japan [1], and only a few have been reported from other countries [5, 6, 15, 21]. Rickets and osteomalacia are common metabolic bone disorders in India [25]. The majority of patients respond to usual therapeutic doses of oral vitamin D and calcium [26], and only a few of them are vitamin D-resistant due to hereditary hypophosphatemia and renal tubular acidosis [27, 28]. In a recent series of 98 patients with rickets/osteomalacia from India, none had VDDR-type II, indicating the rarity of this disease [29].
To date, there have been three reports of VDDR-type II in Asian Indian patients [21, 30, 31]. Sequencing of the VDR gene was carried out in only two cases (sibs), which revealed the presence of a c.776A>C mutation in the LBD [21]. Here, we report a case of VDDR-type II associated with a deletion mutation (c.716delA) in exon 6 of the VDR gene, resulting in a frameshift leading to truncated VDR protein without the LBD, and determine the functional significance of the resultant protein by electrophoretic mobility shift assay (EMSA).
Materials and Methods
Case
A 13-year-old girl, from a northeastern Indian state, presented at the age of 8 years for evaluation of vitamin D-resistant rickets. She was third among five children born to consanguineous parents and was a product of a full-term vaginal delivery with no birth defects. One month following birth there was progressive loss of hair from the scalp and eyebrows. She developed bony pain and bowing of the legs at 1 year of age, for which she was given oral calcium tablets by a local pediatrician. There was no improvement, and she sustained fractures in the left femur and right humerus. Tooth eruption was normal, but she had early tooth decay and extractions. Her elder brother had similar problems and died at the age of six due to pneumonia. Her parents and other siblings were normal.
Her height and weight were 113 cm and 23 kg, respectively, (both <5th percentile). She had sparse scalp hair and bilateral genu valgum (Fig. 1a, b). Investigations revealed serum total calcium of 8.0 mg/dl (normal range 8.1–10.4 mg/dl), inorganic phosphorus of 2.0 mg/dl (normal range 2.5–4.5 mg/dl), alkaline phosphatase of 3,450 IU/l (normal range <18 years 240–840 IU/l), 25(OH)D of 13 ng/ml (normal range 9–37 ng/ml), intact PTH of 353 pg/ml (normal range 15–65 pg/ml), and 1,25(OH)2D of 168 pmol/l (normal range 39–102 pmol/l), as well as 24-h urinary calcium excretion of 10 mg/day, with a urine calcium to creatinine ratio of 0.02. X-rays of the wrist revealed cupping and fraying of the metaphysis, widening of the epiphysis, and generalized osteopenia (Fig. 1c[i]). Baseline arterial blood gas and ammonium chloride loading tests were normal. Skin biopsy showed atrophic epidermis with focal spongiosis and absence of hair shafts. A clinical diagnosis of VDDR-type II was made, and a blood sample was drawn for analysis of VDR mutation after obtaining informed consent from the parents.

a Height of the patient before (i) and after (ii) treatment. b Alopecia before treatment (i) and the mild increase in eyebrow hair after treatment (ii). c X-ray of wrist before treatment showing florid rickets (i) and after treatment showing healing (ii)
During hospitalization, the patient was put on intravenous 10% calcium gluconate infusion (1.4 g elemental calcium/day) along with oral phosphate at a dose of 1 g of elemental phosphorus (neutral phosphate solution, 3.66 g of disodium hydrogen phosphate and 1.0 g of sodium dihydrogen phosphate dissolved in 60 ml). At discharge, she was advised to continue the same dose of calcium infusions and phosphate and cholecalciferol in dose of 60,000 IU/week for the next 8 weeks. Subsequently, she was advised to continue intravenous calcium infusion once a week. On follow-up at 18 months, she showed significant improvement in symptoms and had gained height of 6 cm. There was a mild increase in the eyebrows but no improvement in scalp hair. Serum total calcium (8.7 mg/dl) and inorganic phosphorus (2.4 mg/dl) improved, serum PTH decreased to 52 pg/ml, and serum alkaline phosphatase decreased to 2,223 IU. Repeat radiographs showed signs of healing (Fig. 1c[ii]). She was advised to continue the same treatment. After informed consent, blood was also drawn from the parents and two siblings for estimation of serum total calcium, serum inorganic phosphorus, serum alkaline phosphatase, and serum PTH and for DNA analysis (Fig. 2a). All of them had a normal biochemical profile. Mutation analysis of the VDR was carried out as follows.

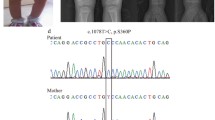
a Pedigree chart. Both parents (F and M) and siblings B1 and S1 were tested for the mutation. Sibling B2 died at the age of 6 years. Solid symbols indicate homozygosity; half-solid symbols indicate heterozygosity; hatched symbols indicate mutation analysis not done. b Electropherogram of exon 6 of the VDR gene from the index patient showing deletion of nucleotide A (c.716delA), shown by down arrow between A and G. c–e Electropherogram of father, mother, and sibling S1, respectively, showing heterozygous deletion of nucleotide A (down arrow between A andG) as predicted by the overlapping peaks. f Electropherogram of sibling B1 showing the wild-type sequence (*)
Mutation Analysis of the VDR Gene Coding Region
Preparation of VDR cDNA from the Affected Patient and a Normal Control for Sequencing
Sequencing of full-length VDR cDNA was performed to look for a mutation in the coding region of the gene in the affected patient and in a normal unrelated healthy Indian subject. VDR cDNA was synthesized using total RNA obtained from peripheral blood mononuclear cells (PBMCs) separated from 10 ml blood using Ficoll paque plus (Amersham, Arlington Heights, IL). The total RNA extracted by RNA binding columns (AG-22331; Eppendorf, Hamburg, Germany) was quantified by spectrophotometer (Genequant, Amersham), and its integrity was checked by the presence of 28S and 18S rRNA bands on 1.5% agarose electrophoresis. First-strand VDR cDNA was synthesized using 2 μg of total RNA, 500 μM dNTPs, 5 μM random hexamers, 20 units of RNase inhibitor, and 200 units of M-MuLV reverse transcriptase (RevertAidTMH Minus; MBI Fermentas, St. Leon-Rot, Germany) in a 20-μl reaction processed at 42°C for 60 min using GenAmp PCR system 9700 (Applied Biosystems, Foster City, CA). The double-stranded full-length VDR cDNA was then amplified using the primers 5′-acctgcccccg g a tccttcagggatg-3′ and 5-accgccaca a gct ttcctagtcagga-3′. Amplification was carried out using high-fidelity proofreading DNA polymerase enzyme (Platinum® Pfx [2.5 U]; Invitrogen, Carlsbad, CA) in 50-μl reaction mixtures containing 5 μl single-stranded cDNA template, 1× PCR buffer, 1 mM MgSO4, 0.3 μM each of the two primers, and 0.3 mM dNTPs, with initial denaturation at 94°C for 7 min, followed by 40 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, extension at 68°C for 90 s, and final extension at 68°C for 15 min. The resultant 1,327-bp product was purified using the QIAEX II gel extraction kit (Qiagen, Hilden, Germany).
Cloning and Sequencing
The 1,327-bp VDR cDNA was cloned for the purpose of sequencing using pPROEX™HTa plasmid expression vector and Escherichia coli DH5α. The nucleotides shown in bold in the set of primers used to prepare double-stranded cDNA indicate the BamHI and HindIII restriction sites, respectively; and the underlined nucleotides indicate the changes introduced in the primers to create the above restriction sites.
Purified cDNA amplicon and pPROEX HTa plasmid expression vector (Invitrogen) were digested with BamHI and HindIII restriction enzymes and ligated using 2.5 units of T4 DNA ligase (MBI Fermentas) in 3:1 ratio (v/v) at 22°C for 6 h. The ligated product was then transformed by the heat shock method into E. coli DH5α-competent cells, which were then grown on LB agar containing 100 μg/ml ampicillin. The successful VDR recombinants were screened using a set of internal primers 5′-gacatcggcatgatgaagg-3′ and 5′-ctagggtcacagaagggtcatc-3′ after dissolving the colonies in 30 μl of 0.01% Triton X-100 and boiling for 20 min using standard PCR conditions. A positive clone was directly sequenced by Big Dye v3·1 Cycle Sequencing Chemistry on the ABI PRISM 310 Genetic Analyzer sequencer (Applied Biosystems) using the primers 5′-agcggataacaatttcacacagg-3′ and 5′-caggctgaaaatcttctctcatc-3′.
Expression and Purification of Wild-Type and Mutant VDR Protein
The plasmids containing wild-type VDR cDNA from a normal individual and VDR cDNA insert prepared from the affected patient were transformed into another E. coli strain, i.e., BL21 (DE3), for protein expression. Two 500-ml cultures of E. coli BL21 (DE3) containing wild-type and mutant VDR plasmid constructs were induced with isopropylthiogalactoside (0.5 mM) and grown for 20 h at 25°C for protein expression. The recombinant proteins were purified by affinity chromatography under nondenaturing conditions from the soluble fraction of E. coli using a Ni-NTA agarose column (Qiagen). The concentration of purified recombinant proteins varied 5.1–5.9 mg/ml. The expression of these proteins was checked by Western blot analysis using a mouse antihistidine tag antibody (1:4,000 dilution; Serotec, Kidlington, UK) and a′ VDR monoclonal antibody (1:10,000 dilution, catalogue V2130X; U.S. Biological, Swampscott, MA) as primary antibodies.
EMSA
EMSA was used to assess the DNA binding ability of the mutant VDR protein. The DNA probes used in EMSA were biotin-labeled oligonucleotide (5′-ttggtgactcaccgggtgaacgggggcatt-3′) and its complementary sequence containing VDR recognition element (underlined). These oligonucleotides containing the VDR element sequence were identical to that present in the human osteocalcin gene [32] and were synthesized using commercial services (MWG, Ebersberg, Germany). The 3′ ends of the two complementary DNA sequences were labeled with biotin in two separate tubes using a commercial kit (Thermo Scientific, Rockford, IL) and then annealed at room temperature for 60 min. In view of the requirement of VDR and retinoid X receptor (RXR) heterodimerization for optimal binding to the vitamin D response element, cloning of the RXR receptor gene and its protein expression was also carried out. Briefly, RXR-α cDNA was prepared from total RNA extracted from the stored duodenal mucosal tissue using gene-specific primers (5′-gccgggcatga a tt cgtcgcagacatg-3′ and 5′-ggtgggcacaaag ct tgggcccgcag-3′). The stored duodenal mucosal biopsy tissue was from a patient with dyspepsia undergoing upper gastrointestinal tract endoscopy and obtained after written informed consent in our earlier study [33]. The nucleotides shown in bold in the set of primers used to prepare double-stranded cDNA indicate the EcoRI and HindIII restriction sites, respectively; and the underlined nucleotides indicate the changes introduced in the primers to create the above restrictions sites. The steps followed for cloning of the RXR-α gene and protein expression were same as described above for VDR protein.
The EMSA reactions were carried out by incubating 4 nmol of DNA probes with either wild-type or mutant VDR protein on ice for 30 min in a 20-μl reaction mixture as described earlier [34, 35]. The reaction contained 120 mM KCl, 20 mM Tris (pH7.5), 1.5 mM EDTA, 2 mM DTT, 5% glycerol, 0.5% CHAPS, 10 mM NaF, 100 μM Na3VO4, 1 μg dIdC (Sigma–Aldrich, St. Louis, MO), and 70 nM 1,25(OH)2D3 (Sigma–Aldrich). RXR-α protein and VDR protein were added in an equimolar ratio in the EMSA reaction. The complete 20-μl reaction mixture was loaded on 6% native PAGE in 0.5 × TBE buffer, transferred to a positively charged nylon membrane (HybondTM-N+; Amersham Biosciences, Aylesbury, UK) at 390 mA for 45 min and cross-linked using a UV cross-linker at 120 mJ/cm2 for 1 min (Amersham). A biotin-labeled DNA probe and its shift were detected using a stabilized streptavidin–horseradish peroxidase conjugate and chemiluminescent substrate luminol (Thermo Scientific) with X-ray film. The density of bands due to DNA protein shift was determined using the Alpha Imager EC (Alpha-Innotech, San Leandro, CA). The specificity of the DNA–VDR–RXR band observed in the EMSA was checked by polyclonal VDR antibody raised in rabbits.
Results
Sequencing of VDR cDNA in the Patient
The nucleotide sequence obtained from the patient was compared with the hVDR complete cDNA sequence (GenBank accession NM_001017535) using the Blastn algorithm (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and revealed the presence of a deletion mutation (c.716delA). This mutation resulted in a frameshift and complete loss of the LBD. To confirm this mutation, sequencing of the genomic DNA region (exon 6) was also carried using primers 5′-cagctctccatgctgccccacctg-3′ and 5′-ggtggatgagtgatctccaacccttc-3′ which amplified a 255-bp PCR product containing the mutated site (Fig. 2b). Analysis of electropherograms obtained from genomic DNA sequencing revealed the presence of a homozygous c.716delA mutation.
Sequencing of Genomic DNA in Relatives for the c.716delA Mutation
Genomic sequencing of exon 6 was also carried out in both parents (F and M) and two siblings (B1 and S1). The parents and S1 revealed a heterozygous form of the same mutation (Fig. 2c–e). The electropherogram of sibling B1 showed a normal wild-type sequence (Fig. 2f).
Figure 3a–d shows the results of EMSA studies carried out to assess the binding ability of mutant and wild-type VDR proteins to the vitamin D response elements of the osteocalcin gene. To determine the optimal dose of VDR protein required to shift the DNA probe in EMSA experiments, a dose–response experiment was carried out using different concentrations of wild-type VDR-RXR protein (24 μg [0.5 nmol], 12 μg [0.25 nmol], and 6 μg [0.12 nmol] per reaction). The results showed that 0.5 nmol of wild-type VDR protein resulted in an appreciable shift of 2 ng of the DNA probe and the presence of two bands due to protein–DNA complexes on EMSA (Fig. 3a, lane 2). Reduction of the wild-type VDR protein concentration to 0.25 nmol and 0.12 nmol in the EMSA reaction resulted in a significant decrease in the intensity of the DNA–protein shift (Fig. 3a, lanes 3 and 4, respectively). Addition of a higher concentration of protein and probe was not possible in our experiments because of the limitation of the EMSA reaction volume and high background on X-ray imaging, respectively. The upper band was due to VDR–RXR heterodimer or VDR homodimer and the lower band was due to the RXR or VDR protein DNA shift. Addition of VDR antibody led to complete disappearance of the upper band, indicating inability of the VDR–RXR–VDR antibody complex to move into the gel in EMSA (Fig. 3b, lane 3). The identity of the upper and lower bands in the assay was further shown by putting EMSA reactions containing either the VDR protein alone or the RXR protein alone. EMSA with RXR protein with no VDR protein showed absence of the upper band but presence of a weak lower band, indicating a DNA protein complex due to the RXR monomer (Fig. 3e, lane 3). In contrast, EMSA with VDR protein alone and no RXR protein showed a prominent upper band due to the VDR homodimer and a weak lower band due to the VDR monomer.

a EMSA showing effect of variability of wild-type VDR protein concentration on DNA binding (lane 1, only DNA probe). Upper arrow shows band due to VDR–RXR–DNA shift or VDR homodimer–DNA shift, and lower arrow indicates the shift due to RXR or VDR monomer. b EMSA showing the specificity of the upper band due to VDR–RXR–DNA shift, as shown by addition of VDR antibodies leading to its complete disappearance due to immobility of the DNA–protein–antibody complex in the PAGE along with reduced intensity of the probe at baseline (lane 3). Lane 2 contains wild-type VDR and RXR in equimolar ratio showing normal shift and presence of upper heterodimer band. Lane 1 contains only DNA probe and arrowhead indicates presence of nonspecific band. c EMSA showing reduced DNA binding ability of mutant VDR protein when added in equimolar ratio with RXR protein (lane 4). Lane 2 contains wild-type VDR and RXR in equimolar ratio showing normal shift. Lane 1 contains only DNA probe. d EMSA showing the effect on increased amount of mutant VDR protein. With an equimolar quantity of RXR protein (0.5 nmol each in lanes 2 and 3), the density of mutated and wild-type VDR–RXR heterodimer achieved comparable values when a two-fold higher concentration of mutated VDR protein (1.0 nmol) was used in the EMSA reaction (lane 3). e EMSA showing the specificity of the upper band due to RXR–VDR heterodimer and the lower band due to RXR or VDR monomer. Lane 2 contains VDR and RXR proteins in equimolar ratio and shows the presence of both upper and lower bands. Lane 3 contains EMSA with RXR protein and no VDR protein, showing absence of the upper band but presence of a weak lower band, indicating DNA protein complex due to RXR monomer. Lane 4 contains EMSA with VDR protein and no RXR protein, showing presence of a prominent upper band due to VDR homodimer and a weak lower band due to VDR monomer. Lane 1 contains only DNA probe, and arrowhead indicates presence of nonspecific band
All EMSA experiments, therefore, were carried out using 0.5 nmol of wild-type full-length VDR protein or an equimolar amount of mutant VDR protein. In the EMSA reaction, the proteins were added in an equimolar amount rather than in equal weight because of differences in length of the amino acid sequence and, hence, the molecular weight of the wild-type and mutant VDR protein. Since 1 nmol of X-kDa protein is equal to X μg of protein, 24 μg of 48-kDa wild-type VDR protein would be equivalent to 0.5 nmol of full-length protein (or ~0.5 nmol of its DBD).
The results of the EMSA experiments indicated that in equimolar concentration the mutant VDR protein had reduced DNA binding ability, as indicated by a reduction in the density of the upper band due to a VDR–RXR–DNA shift, which was 30% less compared to that obtained with wild-type VDR protein (Fig. 3c, lane 2 versus 4). With an equimolar quantity of RXR protein (0.5 nmol each in lanes 2 and 3), the density of the DNA–protein complex obtained with mutated and wild-type VDR–RXR heterodimer achieved comparable values when a two-fold higher concentration of mutated VDR protein (1.0 nmol) was used in the EMSA reaction (Fig. 3d, lane 3). This indicated 50% DNA binding efficacy of the mutant VDR–RXR heterodimer compared to that of the wild-type VDR–RXR heterodimer.
Discussion
The present study reveals the presence of a new deletion mutation, c.716delA, in the LBD of the VDR gene. This deletion mutation resulted in a frameshift, leading to creation of a new stop codon and premature termination of the VDR protein. The mutant protein contains only 246 amino acids instead of the normal 427 amino acids. The first 239 amino acids at the N terminus were of the wild-type sequence, while the subsequent seven amino acids were changed due to the frameshift. Complete loss of the C terminus of the LBD of the mutated VDR protein led to VDDR-type II with severe rickets and alopecia in the present case. The mutation was transmitted in an autosomal recessive manner, and her parents and one of her siblings with the same mutation in heterozygous form were asymptomatic, with no features of metabolic bone disease or alopecia.
Alopecia has been considered one of the indicators of severe hormone resistance in patients with VDDR-type II [36]. Though there is genotype–phenotype variability, alopecia, and severe hormone resistance are common in patients with mutations in DBD, as revealed by the fact that all 11 patients with mutations in the DBD (including nine missense and two nonsense) reviewed by Malloy et al. [1, 7] had severe hormone resistance and severe alopecia/alopecia totalis [see also 8]. In contrast, mutations of the LBD are associated with variable degrees of vitamin D resistance and alopecia [1]. Only 13 of the 21 mutations described to date in the LBD were associated with alopecia (Table 1). Among the eight patients without alopecia, six were resistant to treatment with high doses of vitamin D and required intravenous calcium therapy. The novel mutation described in our patient adds to the variety of LBD mutations with alopecia, which responded partially (improvement in the eyebrows) to intravenous calcium therapy.
The functional significance of mutations in the LBD has been assessed by various investigators (Table 1). The mutated protein results in its inadequate expression [21, 22, 24], defect in the heterodimer formation with RXR protein [5, 9, 21, 23], reduced or absent affinity to the ligand at either the 1-alpha or the 25(OH) region [13, 15, 16, 18, 19, 23], and poor binding of VDR protein to its coactivators [12–14]. Mutations in the LBD which directly affect heterodimerization with RXR result in alopecia [5, 9, 21, 23]. Mutations which affect ligand binding without a defect in heterodimerization are not associated with alopecia [13–15]. Even mutations which completely abolish binding of ligand to VDR may not be associated with alopecia [15, 16, 18]. The severe alopecia noted in our case could possibly be explained by the change in the amino acid sequence 238–246 of the LBD. The VDR domain containing the 241–263 amino acid sequence has been considered important for the interaction between nuclear proteins and RXR heterodimerization [18]. In fact, the EMSA result in the current study indicated 50% reduced DNA binding ability of the VDR–RXR heterodimer.
In view of the complete loss of the LBD and the heterodimerization defect, the present case was treated with intravenous calcium infusion and oral phosphate. This resulted in a significant gain in height and healing of rickets. The healing of rickets with calcium and phosphate therapy without vitamin D indicates passive mineralization of the osteoid if the circulating levels of the minerals are maintained [2]. Despite the healing of the rickets, there was no significant change in the scalp hair. Interestingly, there was mild improvement in the density of the eyebrows.
To conclude, the present report describes the occurrence of a new deletion mutation in the LBD in an Indian patient with VDDR-type II. The mutation resulted in complete loss of the C terminus of the LBD with reduced ability to bind vitamin D response elements. However, rickets partially healed following intravenous calcium and oral phosphate therapy during the course of a year.
References
Malloy PJ, Pike JW, Feldman D (1999) The vitamin D receptor and the syndrome of hereditary 1, 25 dihydroxy vitamin D-resistant rickets. Endocr Rev 20:156–188
Liberman UA, Marx SJ (2003) Vitamin D-dependent rickets. In: Favus MJ (ed) Primer on the metabolic bone diseases and disorders of mineral metabolism, 5th edn. American Society for Bone and Mineral Research, Washington, DC, pp 407–413
Baker AR, McDonnell DP, Hughes M, Crisp TM, Mangelsdorf DJ, Haussler MR, Pike JW, Shine J, O’Malley BW (1988) Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc Natl Acad Sci USA 85:3294–3298
Hughes MR, Malloy PJ, Kieback DG, Kesterson RA, Pike JW, Feldman D, O’Malley BW (1988) Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science 242:1702–1705
Nguyen M, d’Alesio A, Pascussi JM, Kumar R, Griffin MD, Dong X, Guillozo H, Rizk-Rabin M, Sinding C, Bougnères P, Jehan F, Garabédian M (2006) Vitamin D-resistant rickets and type 1 diabetes in a child with compound heterozygous mutations of the vitamin D receptor (L263R and R391S): dissociated responses of the CYP-24 and rel-B promoters to 1,25-dihydroxyvitamin D3. J Bone Miner Res 21:886–894
Miller J, Djabali K, Chen T, Liu Y, Ioffreda M, Lyle S, Christiano AM, Holick M, Cotsarelis G (2001) Atrichia caused by mutations in the vitamin D receptor gene is a phenocopy of generalized atrichia caused by mutations in the hairless gene. J Invest Dermatol 117:612–617
Zhou Y, Wang J, Malloy PJ, Dolezel Z, Feldman D (2009) Compound heterozygous mutations in the vitamin D receptor in a patient with hereditary 1, 25-dihydroxyvitamin D-resistant rickets with alopecia. J Bone Miner Res 24:643–651
Shafeghati Y, Momenin N, Esfahani T, Reyniers E, Wuyts W (2008) Vitamin D-dependent rickets type II: report of a novel mutation in the vitamin D receptor gene. Arch Iran Med 11:330–334
Malloy PJ, Zhu W, Zhao XY, Pehling GB, Feldman D (2001) A novel inborn error in the ligand-binding domain of the vitamin D receptor causes hereditary vitamin D-resistant rickets. Mol Genet Metab 73:138–148
Malloy PJ, Zhu W, Bouillon R, Feldman D (2002) A novel nonsense mutation in the ligand binding domain of the vitamin D receptor causes hereditary 1, 25-dihydroxyvitamin D-resistant rickets. Mol Genet Metab 77:314–318
Arita K, Nanda A, Wessagowit V, Akiyama M, Alsaleh QA, McGrath JA (2008) A novel mutation in the VDR gene in hereditary vitamin D-resistant rickets. Br J Dermatol 158:168–171
Malloy PJ, Wang J, Peng L, Nayak S, Sisk JM, Thompson CC, Feldman D (2007) A unique insertion/duplication in the VDR gene that truncates the VDR causing hereditary 1, 25-dihydroxyvitamin D-resistant rickets without alopecia. Arch Biochem Biophys 460:285–292
Malloy PJ, Xu R, Peng L, Peleg S, Al-Ashwal A, Feldman D (2004) Hereditary 1, 25-dihydroxyvitamin D resistant rickets due to a mutation causing multiple defects in vitamin D receptor function. Endocrinology 145:5106–5114
Malloy PJ, Xu R, Peng L, Clark PA, Feldman D (2002) A novel mutation in helix 12 of the vitamin D receptor impairs coactivator interaction and causes hereditary 1, 25-dihydroxyvitamin D-resistant rickets without alopecia. Mol Endocrinol 16:2538–2546
Malloy PJ, Xu R, Cattani A, Reyes L, Feldman D (2004) A unique insertion/substitution in helix H1 of the vitamin D receptor ligand binding domain in a patient with hereditary 1, 25-dihydroxyvitamin D-resistant rickets. J Bone Miner Res 19:1018–1024
Nguyen TM, Adiceam P, Kottler ML, Guillozo H, Rizk-Rabin M, Brouillard F, Lagier P, Palix C, Garnier JM, Garabedian M (2002) Tryptophan missense mutation in the ligand-binding domain of the vitamin D receptor causes severe resistance to 1, 25-dihydroxyvitamin D. J Bone Miner Res 17:1728–1737
Katavetin P, Katavetin P, Wacharasindhu S, Shotelersuk V (2006) A girl with a novel splice site mutation in VDR supports the role of a ligand-independent VDR function on hair cycling. Horm Res 66:273–276
Kristjansson K, Rut AR, Hewison M, O’Riordan JL, Hughes MR (1993) Two mutations in the hormone binding domain of the vitamin D receptor cause tissue resistance to 1, 25 dihydroxyvitamin D3. J Clin Invest 92:12–16
Malloy PJ, Eccleshall TR, Gross C, Van Maldergem L, Bouillon R, Feldman D (1997) Hereditary vitamin D resistant rickets caused by a novel mutation in the vitamin D receptor that results in decreased affinity for hormone and cellular hyporesponsiveness. J Clin Invest 99:297–304
Hawa NS, Cockerill FJ, Vadher S, Hewison M, Rut AR, Pike JW, O’Riordan JL, Farrow SM (1996) Identification of a novel mutation in hereditary vitamin D resistant rickets causing exon skipping. Clin Endocrinol (Oxf) 45:85–92
Cockerill FJ, Hawa NS, Yousaf N, Hewison M, O’Riordan JL, Farrow SM (1997) Mutations in the vitamin D receptor gene in three kindreds associated with hereditary vitamin D resistant rickets. J Clin Endocrinol Metab 82:3156–3160
Malloy PJ, Hochberg Z, Tiosano D, Pike JW, Hughes MR, Feldman D (1990) The molecular basis of hereditary 1, 25-dihydroxyvitamin D3 resistant rickets in seven related families. J Clin Invest 86:2071–2079
Whitfield GK, Selznick SH, Haussler CA, Hsieh JC, Galligan MA, Jurutka PW, Thompson PD, Lee SM, Zerwekh JE, Haussler MR (1996) Vitamin D receptors from patients with resistance to 1, 25-dihydroxyvitamin D3: point mutations confer reduced transactivation in response to ligand and impaired interaction with the retinoid X receptor heterodimeric partner. Mol Endocrinol 10:1617–1631
Macedo LC, Soardi FC, Ananias N, Belangero VM, Rigatto SZ, De-Mello MP, D’Souza-Li L (2008) Mutations in the vitamin D receptor gene in four patients with hereditary 1, 25-dihydroxyvitamin D-resistant rickets. Arq Bras Endocrinol Metabol 52:1244–1251
Pettifor JM (2007) Vitamin D and/or calcium deficiency rickets in infants and children: a concern for developing countries? Indian Pediatr 44:893–895
Rajeswari J, Balasubramanian K, Bhatia V, Sharma VP, Agarwal AK (2003) Aetiology and clinical profile of osteomalacia in adolescent girls in northern India. Natl Med J India 6:139–142
Batra P, Tejani Z, Mars M (2006) X-linked hypophosphatemia: dental and histologic findings. J Can Dent Assoc 72:69–72
Bajpai A, Bagga A, Hari P, Bardia A, Mantan M (2005) Long-term outcome in children with primary distal renal tubular acidosis. Indian Pediatr 42:321–328
Ray D, Goswami R, Gupta N, Tomar N, Singh N, Sreenivas V (2008) Predisposition to vitamin D deficiency osteomalacia and rickets in females is linked to their 25(OH)D and calcium intake rather than vitamin D receptor gene polymorphism. Clin Endocrinol (Oxf) 71:334–340
Gupta PC, Patwari AK, Mullick DN (1990) Alopecia with rickets: an end organ unresponsiveness to 1, 25-dihydroxyvitamin D—a case report. Indian J Med Sci 44:239–243
Soni SS, Adikey GK, Raman AS (2008) Vitamin D dependent rickets type II: late onset of disease and response to high doses of vitamin D. Saudi J Kidney Dis Transpl 19:796–798
Kerner SA, Scott RA, Pike JW (1989) Sequence elements in the human osteocalcin gene confer basal activation and inducible response to hormonal vitamin D3. Proc Natl Acad Sci USA 86:4455–4459
Goswami R, Mondal AM, Tomar N, Ray D, Chattopadhyay P, Gupta N, Sreenivas V (2009) Presence of 25(OH)D deficiency and its effect on vitamin D receptor mRNA expression. Eur J Clin Nutr 63:446–449
Solomon C, Sebag M, White JH, Rhim J, Kremer R (1998) Disruption of vitamin D receptor-retinoid X receptor heterodimer formation following ras transformation of human keratinocytes. J Biol Chem 273:17573–17578
Koszewski NJ, Reinhardt TA, Langub MC, Malluche HH, Horst RL (1998) Selectivity of a C-terminal peptide antiserum for different DNA-binding states of the vitamin D receptor. Arch Biochem Biophys 349:388–396
Marx SJ, Bliziotes MM, Nanes M (1986) Analysis of the relation between alopecia and resistance to 1, 25-dihydroxyvitamin D. Clin Endocrinol (Oxf) 25:373–381
Thompson E, Kristjansson K, Hughes M (1991) Molecular scanning methods for mutation detection: application to the 1, 25-dihydroxyvitamin D receptor [abstract]. In: Norman AW, Bouillon R, Thomasset M (eds) Vitamin D: gene regulation, structure–function analysis, and clinical application. Eighth Workshop on Vitamin D. Walter de Gruyter, New York, p 6
Acknowledgments
The authors acknowledge the financial help from the Indian Council of Medical Research to N. T. during his senior research fellowship and the help of Dr. Achouba Singh, Senior Resident in the Department of Endocrinology, in the medical management and follow-up of the patient.
Author information
Authors and Affiliations
Corresponding author
Additional information
Jeyaraman Kanakamani and Neeraj Tomar contributed equally to this work.
The authors have stated that they have no conflicts of interest.
Rights and permissions
About this article
Cite this article
Kanakamani, J., Tomar, N., Kaushal, E. et al. Presence of a Deletion Mutation (c.716delA) in the Ligand Binding Domain of the Vitamin D Receptor in an Indian Patient with Vitamin D-Dependent Rickets Type II. Calcif Tissue Int 86, 33–41 (2010). https://doi.org/10.1007/s00223-009-9310-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-009-9310-2