
Abstract
Purpose
Vitamin D-dependent rickets type 1b (VDDR1b) is a very rare autosomal recessive disorder caused by mutations in CYP2R1 that produces 25-hydroxylase. To date only five mutations in CYP2R1 have been identified. This study has reported the genetic results and the clinical characteristics of a family with VDDR1b and compared this family to the other families with VDDR1b in literature.
Methods
After two probands were diagnosed with VDDR1b, all other family members were evaluated. Serum calcium, phosphorus, alkaline phosphatase, parathyroid hormone, 25-hydroxy vitamin D, and 1.25-dihydroxy vitamin D levels were measured in all family members. All individuals were evaluated radiographically, and a genetic analysis was done in all family members. The other families with VDDR1b in literature were reviewed.
Results
Two novel mutations [c.367 + 1G > C and p.E339Q (c.1015G > C)] were identified. The clinic and laboratory findings were strikingly different among the members of this family regardless of the mutation and the number of alleles affected. The families having different mutations in literature had also extensive variation in both the clinical and the laboratory findings.
Conclusion
The current study further expands CYP2R1 mutation spectrum. The findings of both the current and the previous studies suggest that VDDR1b is a more complex disorder than the known autosomal recessive inheritance model and the phenotype may show an extensive variation regardless of the mutation type and the gene dosage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant-derived ergocalciferol (D2) and animal origin cholecalciferol (D3) are exogenous resources of vitamin D (calciferols) in humans. Cholecalciferol can also be produced from the conversion of precholecalciferol that is formed from 7-dehydrocholesterol in the human skin by the solar ultraviolet B radiation at the wavelength of 280–315 nm. Because the extra precholecalciferol is converted by the sunlight to inactive metabolites such as lumisterol and tacisterol, vitamin D intoxication does not happen due to endogen vitamin D production (Fig. 1) [1]. A two-step hydroxylation of carbon atoms at positions 25 and 1 of D2 and D3 is essential to become biologically active vitamin D. The first step happens in the liver where D2 and D3 are hydroxylated to 25-hydroxy D2 and 25-hydroxy D3 by 25-hydroxylase. These products are called 25-hydroxy vitamin D (25OHD). The second step largely occurs in the kidney where 25OHD is hydroxylated to 1.25-dihydroxy vitamin D (1.25 (OH)2D) by the mitochondrial cytochrome P450 enzyme renal 1α-hydroxylase. The biologically active hormone 1.25(OH)2D has a critical role in calcium (Ca) and phosphorus (P) metabolism, bone growth, and cellular differentiation [2]. Renal 1α-hydroxylase, a rate-limiting enzyme, is regulated by serum Ca, P, parathyroid hormone (PTH), and fibroblast growth factor 23 (FGF23). Hypocalcemia, hypophosphatemia, and PTH stimulate renal 1α- hydroxylase, while hypercalcemia, hyperphosphatemia, and FGF23 inhibit it [1, 3, 4]. Another mitochondrial cytochrome P450 enzyme 24-hydroxylase converts 25OHD and 1.25(OH)2D to inactive metabolites such as 24.25 (OH)2D and 1.24.25(OH)3D, respectively. In addition to this classic pathway, 25OHD and 1.25(OH)2D can also be produced by some alternative pathways especially in the case of 25-hydroxylase or renal 1α-hydroxylase deficiency (Fig. 1).

The classic and alternative pathways of vitamin D metabolism
Vitamin D-binding protein (VDBP), a serum α2-globulin, is responsible for binding and transporting of all vitamin D metabolites such as D2, D3, 25OHD, 1.25(OH)2D, 1-OH-D3, 24.25(OH)2D, and 1.24.25(OH)3D (Fig. 1). This is important because the half-life of free serum vitamin D is very short. Certain polymorphisms in VDBP gene can alter serum vitamin D status by affecting VDBP–vitamin D binding [5].
The clinical picture that develops due to the deficiency or ineffectiveness of 25OHD or 1.25(OH)2D is defined as rickets. Rickets is basically divided into two groups as calciopenic rickets and phosphopenic rickets. Clinical manifestations such as craniotabes, craniosynostosis, frontal bossing, delayed closure of the anterior fontanel, delayed eruption of the teeth, dental abscesses, chest deformities, rachitic rosaries, Harrison sulcus, widening of the wrists, leg deformities, and waddling gait may show various combinations according to the age and different types of rickets. Hypophosphatemia, high serum alkaline phosphatase (ALP) level, and radiologic findings such as enlargement of the epiphysis and cupping/fraying in the metaphysis on the wrist and knee radiographs, and genu valgum or varum in the lower extremity radiographs are in common findings of calciopenic and phosphopenic rickets. However, calciopenic rickets has normal/low serum Ca, secondary hyperparathyroidism, normal/low serum 25OHD, variable serum 1.25(OH)2D, variable urine P, and low urine Ca levels, while phosphopenic rickets has normal/high serum Ca, normal serum PTH, normal serum 25OHD, variable 1.25(OH)2D, high urine P, and variable urine Ca levels (Table 1) [6].
Vitamin D-dependent rickets type 1b (VDDR1b) (OMIM #600081), a genetic form of calciopenic rickets, is a very rare autosomal recessive disorder caused by mutations in CYP2R1 that produces 25-hydroxylase. To date only five mutations in CYP2R1 have been identified (Fig. 2). This disorder is characterized by hypocalcemia, secondary hyperparathyroidism, and low serum concentrations of 25OHD, with insufficient clinical and biochemical responsiveness to conventional doses of vitamin D [7,8,9,10,11].

The previous and novel mutations in CYP2R1. All boxes demonstrate the exons of CYP2R1. The novel mutations in the current study are highlighted in red
In this article, we have reported the genetic results and the clinical characteristics of a family with VDDR1b and identified two novel mutations in CYP2R1. We have also compared this family to the other families with VDDR1b in literature. The findings of both the current and the previous studies suggest that VDDR1b is a more complex disorder than the known autosomal recessive inheritance model and the phenotype may show an extensive variation regardless of the mutation type and the gene dosage.
Method
Patients
Two sisters, aged 16 and 14 years, presented with paresthesia, bone pain, and muscle cramps in their hands and legs. They were the first and second child (II-1 and II-2 in Fig. 3) of a non-consanguineous Turkish couple (I-1 and I-2 in Fig. 3). The old sister (patient II-1) developed a dislocation in her arm at age 6 years, while the young sister (patient II-2) had a bone fracture in her arm at 5 years. Both probands had normal developmental milestones, and the family history for metabolic bone disease was unremarkable.

Pedigree tree of the family.  = partly affected homozygous individual;
= partly affected homozygous individual;  = affected heterozygous individual;
= affected heterozygous individual;  = affected compound heterozygous individuals;
= affected compound heterozygous individuals;  = non-affected heterozygous carrier; wt wild type
= non-affected heterozygous carrier; wt wild type
One year ago both children were hospitalized in another institution due to hypocalcemia. They were both given bolus D3 therapy at different doses and time intervals after the admission.
They both wore Muslim-style covered clothing suggesting inadequate sun exposure and had no other drug history. The daily calcium intake of the patients was calculated according to the 3-day food record.
Siblings and parents
The 42-year-old father (I-1 in Fig. 3) and all other siblings (II-3, II-4, II-5 in Fig. 3) had no any symptoms or a history of bone deformities.
The 44-year-old mother (I-2 in Fig. 3) was receiving medical therapy for hyperthyroidism. Her family history was negative for skeletal deformities. She was given three times bolus 300,000 IU D3 therapy at different time intervals because of leg pain and low serum 25OHD level. She was already using 1000 IU/day D3.
The written consent was obtained from parents after full explanation of the purpose and nature of all procedures used. Ethical approval for this study was obtained from Ataturk University Ethics Committee.
Biochemical measurements
Serum Ca, P, ALP levels were measured with the Boehringer Mannheim-Hitachi System by manufacturer’s protocol. A two-site immunoradiometric assay was used to measure serum PTH concentration. Serum 25OHD levels were measured by the LIAISON® 25-OH Vitamin D TOTAL chemiluminescent immunoassay from DiaSorin, Germany according to the manufacturer’s protocol. Serum 1.25(OH)2D concentrations were measured in serums samples by the DIA source 1.25(OH)2-VIT.D-RIA-CT Kit from DIA source ImmunoAssays S.A, Belgium according to the manufacturer’s protocol.
Radiologic evaluation
Plain radiographs of the extremities and columna vertebralis were obtained in all family members. The wrists and knees radiographs were evaluated for The Rickets Severity Score (RSS) according to metaphyseal fraying, concavity, and the proportion of the growth plate affected by the same radiologist at the diagnosis and follow-up [12].
Molecular biology
Genomic DNA was isolated from peripheral blood leukocytes using a QIAamp DNA Blood Mini QIAcube Kit (Qiagen, Hilden, Germany), according to the manufacturers’ protocols. All coding exons and exon-intron boundaries of the CYP2R1 (Refseq number: NM_024514) were amplified using custom design primers. All PCR products were sequenced via ABI PRISM® 3130xl genetic analyzer (Applied Biosystems, Foster City, CA, USA).
In order to evaluate the pathogenicity of the novel variants, we used in silico prediction tools, mutation databases (Human Gene Mutation Database and Clinvar), allel frequency in population studies (1000 Genome, Genome Aggregation Database (gnomad)), segregation analysis, and American College of Medical Genetics and Genomics criteria [13].
Results
Patients’ clinical and laboratory characteristics
The 3-day food record revealed that both patients had a calcium intake of 600 mg/day. Physical examination findings of both patients were normal except for mild genu valgum. The body weight and height measurements and BMI values of patient II-1 and patient II-2 were 46.8 kg (−1.6 SDS) and 42.5 kg (−1.7 SDS); 158.1 cm (−0.7 SDS) and 157 cm (−0.5 SDS); and 18.7 kg/m2 (−1.2 SDS) and 17.2 kg/m2 (−1.5 SDS), respectively (Table 2).
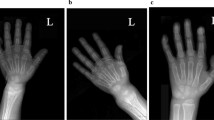
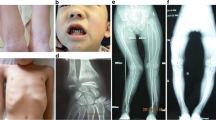
One year before the diagnosis, both patients had hypocalcemia, an elevated ALP level, hyperparathyroidism, and a low 1.25(OH)2D concentration (Table 2). At the time of diagnosis, biochemical evaluation revealed 25OHD deficiency in patient II-1, elevated 1.25(OH)2D and ALP levels in patient II-2, and hyperparathyroidism in both of them (Table 2). Serum P and ALP levels were evaluated according to age and age- and sex-specific reference ranges, respectively. Radiologic evaluation showed genu valgum in both patients (Fig. 4) and the RSS values were 2 and 0 at the diagnosis and 5 months after treatment, respectively.

Genu valgum deformity in patient 1
Siblings and parents’ clinical and laboratory characteristics
Except for a low body weight and an elevated serum ALP level in one sibling (II- 3 in Fig. 3), all clinical assessments and laboratory data were normal in all other siblings. Only that sibling’s characteristics are shown in Table 3.
The father’s (family I in Table 3 and family I-1 in Fig. 3) serum 25OHD level was consistent with vitamin D deficiency, while other biochemical parameters were normal. He was referred to the internal outpatient clinics and given a bolus 300,000 IU D3 therapy.
The mother’s (family I in Table 3 and family I-2 in Fig. 3) biochemical findings were normal except for mild hyperparathyroidism.
Sequence analysis of CYP2R1
The molecular analysis of CYP2R1 identified two novel variant [c.367 + 1G > C and p.E339Q (c.1015G > C)] in a compound heterozygous state in both probands and two other siblings (II-1, II-2, II-3, II-5 in Figs. 3 and 5). They were in intron 2 and exon 4, respectively (Fig. 2). The father was homozygous for c.367 + 1G > C, while the mother was heterozygous for p.E339Q (c.1015G > C). One sibling (II-4 in Fig. 3) had a heterozygous mutation for c.367 + 1G > C.

In affected siblings, A heterozygous paternal c.367 + 1G > C and B heterozygous maternal p.E339Q (c.1015G > C) mutations were detected in intron 2 and exon 4 of the CYP2R1, respectively. C In the mother, heterozygous p.E339Q (c.1015G > C) mutation was detected in exon 4 of the CYP2R1. D In the father, homozygous c.367 + 1G > C mutation was detected in intron 2 of the CYP2R1
These new variants were absent in the gnomad and their site was a highly conserved region among the species. To predict the mechanistic consequences of the variants, in silico analysis was carried out and it showed that the first variant c.367 + 1G > C has great impact on the structure and function of the 25-hydroxylase. The G to C transition in the first nucleotide of the highly conserved splice donor sequence of intron 2 probably leads to improperly spliced mRNA that encodes truncated CYP2R1 proteins or unstable mRNA. The second variant [p.E339Q (c.1015G > C)] was detected in exon 4. Segregation analysis showed that p.E339Q could segregate with the disease phenotype by causing the polar acidic amino acid to turn to the neutral one.
Treatment and follow-up
Patient II-1 was treated with D3 that led to an elevated level of 25OHD and a reduced level of PTH and ALP. Patient II-2 was not given any medicine at the diagnosis because her serum 25OHD and 1.25(OH)2D levels were not low. One month later, however, D3 therapy was started because her serum 25OHD level was low. The patients were followed monthly, and D3 dose was arranged by their serum 25OHD levels. The aims of the treatment were to keep serum 25OHD level within normal limits and to maintain the trend of resolving in biochemical markers. Five months later, this therapy resulted in disappearance of the symptoms and normalization of all biochemical parameters (Table 2).
Discussion
In this study, we increased the number of CYP2R1 mutations responsible for VDDR1b to seven by identifying two new mutations in a family. We have also compared this family to the other families with VDDR1b in literature. All findings suggest that VDDR1b is a more complex disorder than the known autosomal recessive pattern of inheritance and that phenotype may show wide variation regardless of mutation type and gene dosage.
Distinctive biochemical features of VDDR1b from other rickets forms
Rickets is basically divided into two groups as calciopenic rickets and phosphopenic rickets. Serum PTH level is very important for differential diagnosis because it is high in calciopenic rickets and normal in phosphopenic rickets (Table 1). We focused on calciopenic rickets because our patients had secondary hyperparathyroidism. Vitamin D deficiency and VDDR1b are forms of calciopenic rickets characterized by low serum 25OHD level. At this point, the reduced clinical and biochemical responsiveness to the therapeutic dose(s) of vitamin D is the most helpful finding for VDDR1b [7,8,9,10,11]. Although our patients received bolus D3 therapy at different time intervals for 1 year, secondary hyperparathyroidism was still present at the time of diagnosis and the serum 25OHD level of patient II-1 was low. (Table 2). Therefore, VDDR1b was accepted as possible diagnosis in our patients. Since their biochemical profile and unresponsiveness to vitamin D therapy were against phosphopenic rickets, urinary P and Ca excretions were not studied.
Conventional radiography
The RSS is a quantitative method, validated in nutritional rickets, to assess rickets severity in the wrists and knees based on the degree of metaphyseal fraying, concavity, and the proportion of the growth plate affected [6]. It is a ten-point scale, where 10 represents the most extreme degree of rickets severity and 0 represents the absence of radiographic changes of rickets. The radiographic response following treatment of nutritional rickets can be assessed by the RSS, and RSS values correlate with values of serum ALP, a biochemical measure of rachitic activity. The RSS is a reliable and valid instrument for evaluating the severity of radiographic rickets. In our patients, the RSS values were 2 and 0 at the diagnosis and 5 months after treatment, respectively, which was consistent with the ALP decline trend. These findings show that our patients were treated successfully.
Bone formation and resorption markers
Bone consists mainly of bone matrix and mineralization. Rickets refers to insufficient mineralization of the growth plate in the pediatric population. In contrast, osteomalacia refers to insufficient mineralization of the bone matrix that can occur in adults even after the growth plate is closed. Although rickets and osteomalacia are characterized by increased bone turnover, there is no imbalance between bone formation and bone resorption. Therefore, bone mass is not significantly affected [14]. On the other hand, osteoporosis refers to a decrease in both bone matrix and mineralization with changes in bone quality or structure. It is characterized by low bone mass due to an imbalance in favor of bone resorption over bone formation [15]. There are many methods to evaluate bone mass and bone formation/resorption. Dual-energy x-ray absorptiometry (DEXA) is the most useful, easy, and inexpensive method to measure bone mass in children. Bone-specific ALP, osteocalcin, and N-terminal and C-terminal propeptide of type I collagen are biochemical markers of bone formation, while N-terminal and C-terminal telopeptide of type I collagen, tartrate-resistant acid phosphatase 5b, and pyridinoline cross-links are biochemical markers of bone resorption [16]. Among these parameters, serum ALP level is the most important biochemical marker showing negative correlation with bone mass in both children and adults [17,18,19]
Children who have primary bone disorders such as idiopathic juvenile osteoporosis and osteogenesis imperfecta; chronic inflammatory disorders; chronic immobilization; endocrine disorders; cancers and therapies; hematologic disorders; some genetic disorders (e.g., Ehlers Danlos syndrome, galactosemia, and Marfan syndrome); and potentially bone-toxic drugs (e.g., glucocorticoids, methotrexate, and anticonvulsants) are at risk of osteoporosis. If these children have recurrent fractures, bone pain, bone deformities, or “osteopenia” (a term describing the appearance of “washed out” bones) on conventional radiographs, DEXA scans are recommended to identify those at greatest risk of skeletal fractures, to guide decisions regarding treatment, and to monitor responses to therapy [20]. Our patients were diagnosed with VDDR1b not osteoporosis and there was no predisposing factor for low bone mass. Additionally, it should also be noted that interval growth changes and accompanying increases in bone size make it more difficult to differentiate true increases in density from changes in bone mass that are related to growth [20]. Therefore, the DEXA scans were not performed and bone resorption markers were not measured. The recovery in bone mass was monitored by serum ALP level.
The known mutations and novel variants in CYP2R1
To date, five mutations in CYP2R1 encoding 25-hydroxylase have been identified. They are p.L99P (c.296T > C) [7, 8, 18], c.367 + 1G > A, p.L257Sfs*6 (c.768dup) [9], p.K242N (c.726A > C) [10], and p.G42_L46delinsR (c.124_138delinsCGG) [11]. S.J. Casella et al. [7] first reported two Nigerian siblings who had severe rickets despite a history of adequate vitamin D intake. Treatment with high doses of D2 resulted in resolution of the biochemical abnormalities and radiographic deformities. Both patients needed repeated pharmacologic doses of D2 to keep their 25OHD level in normal limits even though vitamin D absorption was normal. So, authors speculated that there might have an inherited defect in conversion of vitamin D to 25OHD. J.B. Cheng et al. [8] showed that one sibling had a homozygous mutation (p.L99P) in CYP2R1. Q. Dong and W.L. Miller [21] confirmed the same mutation in other sibling as well. A.N. Al Mutair et al. [9] reported two Saudi Arabian siblings who had a compound heterozygosity for two new mutations (c.367 + 1G > A and p.L257Sfs*6) [9]. Then, T.D. Thacher et al. [10] identified another new mutation (p.K242N) in combination with a previous one (p.L99P) in two Nigerian siblings. Finally, A. Molin et al. [11] discovered a different mutation (p.G42_L46delinsR) in a French patient (Fig. 2). According to in vitro analysis methods performed, all reported mutations have been predicted to result in nonfunctional proteins, but neither molecular nor functional studies have been presented to confirm these predictions [22]. In the present study, molecular analysis of genomic DNA revealed that both probands had compound heterozygosity for previously undescribed two variants in CYP2R1. The first novel variant (c.367 + 1G > C) was absent in gnomad and had the change of a different base at the same acceptor site previously reported by A.N. Al Mutair et al. [9]. This finding suggests intron 2 is a susceptible site to mutational changes in CYP2R1. The second variant [p.E339Q (c.1015G > C)] was also absent in gnomad. In silico and segregation analyses revealed that these variants were pathogenic and could segregate with the disease phenotype. To the best our knowledge, our patients are the first cases with VDDR1b resulted from two novel mutations (p.E339Q and c.367 + 1G > C) of CYP2R1.
The genotype–phenotype relationship in homozygous and compound heterozygous mutations of CYP2R1
In this study, we have reviewed the clinical and laboratory characteristics in the patients with VDDR1b (Tables 3 and 4). We have realized that the clinical picture is very heterogeneous from only laboratory abnormalities to severe rickets by depending on the patient’s age or genetic mutation. Walking delay, bone pain, bone deformity, and short stature are the most common clinical presentations in children with VDDR1b, and the most severe phenotype seems to be associated with a homozygous p.L99P and a compound heterozygous c.367 + 1G > A and p.L257Sfs*6 mutations (families II, III, IV, and VII in Table 4; the siblings in family VII in Table 3). Our cases had a moderate phenotype characterized by hypocalcemia requiring hospitalization and mild genu valgum in the lower extremities. Interestingly, the genetic studies of the families in literature have revealed some striking results. A 49-year-old adult (the father of family IV in Table 4 and family IV in Table 3) only had a low vitamin D level, while two siblings and three adults from the same family (the relatives of family VII in Table 4 and the siblings, aunts, and uncle in family VII in Table 3) had a less severe bone disease although all of them were homozygous for L99P mutation. Similarly, although our patients’ father (I-1 in Fig. 3) was homozygous for c.367 + 1G > C, he only had a low serum 25OHD concentration (family I in Table 3). More interestingly, although two siblings (II-3 and II-5 in Fig. 3) had the same compound heterozygous variants, sibling II-3 did not have any abnormality except for low body weight and an elevated serum ALP level, while sibling II-5 was completely normal. These findings are very similar to the natural course of many patients with VDDR2A, which have very heterogeneous course even if they have the same mutation. Additionally, all findings suggest that the phenotype can improve with age thanks to the acquisition of a vitamin D independent mechanism for intestinal absorption of calcium due to the elevation of sex hormones with puberty [23, 24]. Alternatively, CYP27A1 and CYP2J2 can produce 25OHD from D3 and D2, respectively, while CYP3A4 can generate 1.25(OH)2D from 1α-OHD (Fig. 1) [25,26,27,28]. These alternate enzymes that possess 25-hydroxylase activity seem to have a greater effect with maturation, and they may also explain why some family members have normal or high serum 1.25(OH)2D concentrations (Tables 3 and 4).
The genotype–phenotype relationship in heterozygous mutations of CYP2R1
VDDR1b is inherited in an autosomal recessive manner. Heterozygous family members for p.G42_L46delinsR (family VI in Table 3), as expected, had no any abnormal clinic or laboratory findings, while the heterozygous members for p.K242N and p.L99P (family V in Table 3 and family V in Table 4, respectively) interestingly had a less severe phenotype. In the present study, our patients’ mother (family I, I-2 in Table 3) also had a milder phenotype despite heterozygosity for the p.E339Q mutation. Given that the father with the homozygous c.367 + 1G > C mutation did not have any clinical abnormalities, the findings in our patients suggest that the maternally derived p.E339Q mutation may have a greater effect on the disease phenotype. All these findings imply VDDR1b is a more complex disorder than the known autosomal recessive inheritance model. However, some other factors such as VDBP, Ca intake, and vitamin D status may have interacted with clinical heterogeneity. The polymorphisms rs4588 and rs7041 in VDBP gene have been found to be strongly associated with differences in serum vitamin D status [5]. This finding suggests that VDBP can affect serum vitamin D level. On the other hand, according to the 1989 Recommended Dietary Allowance, the recommended daily Ca intake for adolescents is 1200 mg/day. However, average Ca retention is only 57% of this amount. Comparing Ca balance in girls with very low (400 mg/day) and higher Ca intake (1200–1300 mg/day) showed little adaptation with increased absorption and decreased excretion in girls with low intakes, but this adaptation was not adequate to account for the large gap in intakes. The net Ca balance was much lower in the girls with low intake. This suggests that intake of <500 mg/day can lead to large Ca deficits [29]. The Ca intake in our patients was 600 mg/day, suggesting that they had a large borderline calcium deficit. This finding implies that insufficient Ca intake may contribute to clinical heterogeneity in our patients. In addition, the fact that most of the families with VDDR1b in the literature are from Africa and the Middle East and some Nigerian patients are treated with only calcium supplementation support our idea [10, 30].
Treatment in patients with VDDR1b
Several options in the treatment of VDDR1b are possible. Because this disorder is related to the defect in 25-hydroxylation of D2 and D3, the use of calcifediol (25-OH-D3) in treatment, bypassing 25-hydroxylation, seems a rational and more physiologic approach. A. Molin at al. [11] used calcifediol in two patients and achieved the normalization in all biochemical parameters and the correction in long bone deformations without developing hypercalcemia (families VI and VII in Table 4). However, calcifediol is a more expensive therapy than D2 or D3 and not available in most countries. At this point, D2 or D3 can be an alternative therapy thanks to CYP27A1 and CYP2J2 that can produce 25OHD. Furthermore, CYP27A1 has the potential to convert 1α-hydroxy D3 to 1.25(OH)2D (Fig. 1). The findings from literature review suggest that the D2 or D3 therapy can normalize plasma concentrations of 25OHD and restore biochemical and radiologic abnormalities (Table 4). Additionally, the serum 1,25(OH)2D level in the patients with VDDR1b is usually normal or high (Table 4). So, the therapies such as calcitriol (1.25(OH)2D3) or alfacalcidol (1α-(OH)D3) should not be preferred because they may cause hypercalcemia because of a rapid elevation of serum 1.25(OH)2D level.
Data on the optimal dosage of vitamin D preparations in VDDR1b patients are very limited in the literature. This is because VDDR1b is a very rare disease and there are no randomized clinical trials. Different therapeutic doses and bolus dose ranges have been reported in case reports or small case series (7, 9–11). We thought that the optimal D3 dose was the amount of D3 that kept the serum 25OHD level within normal limits and continued the improvement trend in biochemical markers. Therefore, we followed the patients with frequent visits to determine the optimum bolus D3 dose and interval time although this approach was not universally accepted. They were successfully treated with D3 without hypercalcemia and their biochemical findings were completely normal 5 months after therapy. We think that bolus D3 dose and interval time should be individualized in patients with VDDR1b.
Conclusion
Thanks to this family, this article expands the number of CYP2R1 mutations responsible for VDDR1b to 7. The findings of both the current and the previous studies suggest that VDDR1b is a more complex disorder than the known autosomal recessive inheritance model, and that phenotype may show an extensive variation regardless of the mutation type and the gene dosage.
Data availability
Some or all data generated or used during the study are available from the corresponding author by request.
References
M.F. Holick, Vitamin D deficiency. N. Engl. J. Med. 357(3), 266–281 (2007)
C.J. Kim, L.E. Kaplan, F. Perwad, N. Huang et al., Vitamin D 1alpha-hydroxylase gene mutations in patients with 1alpha-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 92(8), 3177–3182 (2007)
W.L. Miller, A.A. Portale, Vitamin D biosynthesis and vitamin D 1 alpha-hydroxylase deficiency. Endocr. Dev. 6, 156–174 (2003)
S. Liu, L.D. Quarles, How fibroblast growth factor 23 works. J. Am. Soc. Nephrol. 18(6), 1637–1647 (2007)
D. Rozmus, A. Ciesielska, J. Płomiński et al., Vitamin D binding protein (VDBP) and its gene polymorphisms—the risk of malignant tumors and other diseases. Int. J. Mol. Sci. 21(21), 7822 (2020)
T.O. Carpenter, N.J. Shaw, A.A. Portale et al., Rickets. Nat. Rev. Dis. Primers. 21, 17101 (2017)
S.J. Casella, B.J. Reiner, T.C. Chen et al., A possible genetic defect in 25-hydroxylation as a cause of rickets. J. Pediatr. 124(6), 929–932 (1994)
J.B. Cheng, M.A. Levine, N.H. Bell et al., Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc. Natl Acad. Sci. USA 101(20), 7711–7715 (2004)
A.N. Al Mutair, G.H. Nasrat, D.W. Russell, Mutation of the CYP2R1 vitamin D 25-hydroxylase in a Saudi Arabian family with severe vitamin D deficiency. J. Clin. Endocrinol. Metab. 97(10), E2022–E2025 (2012)
T.D. Thacher, P.R. Fischer, R.J. Singh et al., CYP2R1 mutations impair generation of 25-hydroxyvitamin D and cause an atypical form of vitamin D deficiency. J. Clin. Endocrinol. Metab. 100(7), E1005–E1013 (2015)
A. Molin, A. Wiedemann, N. Demers et al., Vitamin D-dependent rickets type 1B (25-hydroxylase deficiency): a rare condition or a misdiagnosed condition? J. Bone Miner. Res. 32(9), 1893–1899 (2017)
T.D. Thacher, J.M. Pettifor, P.J. Tebben et al., Rickets severity predicts clinical outcomes in children with X-linked hypophosphatemia: utility of the radiographic Rickets Severity Score. Bone 122, 76–81 (2019)
S. Richards, N. Aziz, S. Bale et al., Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17(5), 405–424 (2015)
R.M. Shore, R.W. Chesney, Rickets: part I. Pediatr. Radiol. 43(2), 40–51 (2013)
P. Lips, N.M. van Schoor, The effect of vitamin D on bone and osteoporosis. Best. Pract. Res. Clin. Endocrinol. Metab. 25(4), 585–591 (2011)
S. Jain, P. Camacho, Use of bone turnover markers in the management of osteoporosis. Curr. Opin. Endocrinol. Diabetes Obes. 25(6), 366–372 (2018)
J.M. Pettifor, G.P. Moodley, Appendicular bone mass in children with a high prevalence of low dietary calcium intakes. J. Bone Miner. Res. 12(11), 1824–1832 (1997)
S. Tariq, S. Tariq, K.P. Lone et al., Alkaline phosphatase is a predictor of Bone Mineral Density in postmenopausal females. Pak. J. Med. Sci. 35(3), 749–753 (2019)
Y.-X. Jiang, S.-Y. Tang, X.-P. Wu, et al., Biochemical markers of age-related changes in bones turnover and bone mineral density in healthy Chinese men. Zhong Nan Da Xue Xue Bao Yi Xue Ban 33(1), 53–56 (2008).
L.K. Bachrach, C.M. Gordon, Bone densitometry in children and adolescents. Pediatrics 138(4), e20162398 (2016)
Q. Dong, W.L. Miller, Vitamin D 25-hydroxylase deficiency. Mol. Genet. Metab. 83(1-2), 197–188 (2004)
M.A. Levine, Diagnosis and management of vitamin D dependent rickets. Front. Pediatr. 8, 315 (2020)
D. Tiosano, S. Hadad, Z. Chen et al., Calcium absorption, kinetics, bone density, and bone structure in patients with hereditary vitamin D-resistant rickets. J. Clin. Endocrinol. Metab. 96(12), 3701–3709 (2011)
S.J. Van Cromphaut, K. Rummens, I. Stockmans et al., Intestinal calcium transporter genes are upregulated by estrogens and the reproductive cycle through vitamin D receptor-independent mechanisms. J. Bone Miner. Res. 18(10), 1725–1736 (2003)
G. Jones, S.A. Strugnell, H.F. DeLuca, Current understanding of the molecular actions of vitamin D. Physiol. Rev. 78(4), 1193–1231 (1998)
G. Jones, D.E. Prosser, M. Kaufmann, Cytochrome P450-mediated metabolism of vitamin D. J. Lipid Res. 55(1), 13–31 (2014)
J. Zhu, H.F. de Luca, Vitamin D 25-hydroxylase-four decades of searching, are we there yet? Arch. Biochem. Biophys. 523(1), 30–36 (2012)
J.B. Cheng, D.L. Motola, D.J. Mangelsdorf et al. De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxilase. J. Biol. Chem. 278(39), 38084–38093 (2003)
S.A. Abrams, I.J. Griffin, P.D. Hicks et al. Pubertal girls only partially adapt to low dietary calcium intakes. J. Bone Miner. Res. 19(5), 759–763 (2004)
T.D. Thacher, P.R. Fischer, J.M. Pettifor, Vitamin D treatment in calcium-deficiency rickets: a randomised controlled trial. Arch. Dis. Child. 99(9), 807–811 (2014)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ozden, A., Doneray, H. & Turkyilmaz, A. Two novel CYP2R1 mutations in a family with vitamin D-dependent rickets type 1b. Endocrine 72, 852–864 (2021). https://doi.org/10.1007/s12020-021-02670-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-021-02670-9