
Abstract
Whether antisaccade errors in schizophrenia are due to defects in implementing saccadic inhibition or difficulty in generating novel responses is uncertain. We investigated whether antisaccade errors were related to difficulty in inhibiting saccades when subjects were asked to maintain steady fixation, a situation that does not require a novel response. We examined the ocular motor data of 15 schizophrenia subjects and 16 healthy subjects. We assessed fixation in two situations: first, during the period before target onset during each saccadic trial, and second, during fixation trials that were interspersed with saccadic trials. We found that schizophrenia subjects had higher rates of fixation losses than control subjects in both situations. Second, both in healthy and schizophrenia subjects, antisaccade error rate was positively correlated with the frequency of fixation losses in the preparatory period of saccadic trials, but not with the frequency of fixation losses during fixation trials. Third, antisaccade errors were more likely to occur in trials with unstable fixation than in trials with stable fixation. Last, antisaccade error rate was also correlated with prosaccade error rate. We conclude that antisaccade errors are related to difficulties with implementing inhibitory control in the saccadic system. However, the finding of a correlation between the error rates for antisaccades and prosaccades suggests that this is not specifically concerned with inhibiting the automatic prosaccade, but a more general deficit in implementing goal-oriented behavior.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the most consistent cognitive deficits in schizophrenia is the increased errors on the antisaccade task (for review, see Hutton and Kennard 1998; Broerse et al. 2001; Reuter and Kathmann 2004; Hutton and Ettinger 2006). The antisaccade requires subjects to suppress the habitual tendency to look directly at a suddenly appearing visual target (a prosaccade), and instead make an eye movement towards a different location, traditionally one equidistant from fixation but 180° opposite to the visual stimulus (Hallett and Adams 1980). As such, the antisaccade is a paradigmatic example of the exercise of volitional control over our actions: to perform it correctly, one must execute a novel response instead of a more automatic one. A large body of literature has consistently shown that schizophrenia subjects have a high error rate on this task, ranging from 30 to 70% compared to 10% for healthy controls (Hutton and Ettinger 2006), and longitudinal studies have shown good test–retest consistency of antisaccade error rates over several years of follow-up, with r values of 0.73–0.87 (Calkins et al. 2003; Gooding et al. 2004). This stability suggests that deficient antisaccade performance is a robust marker of a schizophrenic trait, rather than of a disease state. Although there remains debate about whether there is an increased rate of antisaccade errors in relatives of subjects with schizophrenia (Calkins et al. 2004; Levy et al. 2004), antisaccade deficits have been proposed as a possible “endophenotype” (Gottesman and Gould 2003) of schizophrenia that may clarify patterns of inheritance in genetic studies (Radant et al. 2006). However, for the antisaccade deficit to be optimally useful in genetic studies we need to identify exactly which processes fail.
Antisaccade abnormalities are not attributed to defects in the machinery for saccadic generation, since most studies report that in schizophrenia the latencies of simple prosaccades performed in single-task blocks are normal (Fukushima et al. 1990a; Radant et al. 1997; Maruff et al. 1998; Nieman et al. 2000; Hutton et al. 2002) or even slightly faster (Levy et al. 1998). (Error rate for prosaccades is usually negligible and often not reported, though one recent study claims that schizophrenia subjects make more prosaccade errors (Boudet et al. 2005).) Rather, it is suggested that the increased antisaccade error rate in schizophrenia subjects is due to a deficit in prosaccade suppression, and therefore with inhibition (Levy et al. 1998). Neurophysiologic studies of the frontal eye field and superior colliculus show that error rate is inversely correlated with the level of preparatory neural activity in the period immediately prior to stimulus appearance (Everling et al. 1999; Everling and Munoz 2000). An instruction to make an antisaccade reduces the baseline neural firing rate during this period, presumably making it more difficult for activity related to an unwanted prosaccade to reach the threshold for triggering an eye movement. The greater the reduction, the lower the likelihood of an error. However, the price for this improved accuracy is that the activity generating the correct antisaccade response takes longer to reach the threshold, resulting in longer saccadic latencies. This finding thus provides a physiologic basis for speed–accuracy trade-offs in the ocular motor system.
Therefore, if an increase in antisaccade errors occurs through a failure to reduce preparatory activity in these structures, then one should also find shorter latencies for correct antisaccades. This has not been reported in schizophrenia subjects. Rather, many studies find longer antisaccade latencies (Fukushima et al. 1990b, 1994; Müller et al. 1999; Manoach et al. 2002). This combination of increased antisaccade errors and increased latencies of correct antisaccades in schizophrenia suggests that the deficit does not lie in a specific failure to inhibit preparatory activity in the frontal eye field or superior colliculus during antisaccade trials, and that other explanations must be sought.
One possibility is that there is a failure not in some specific inhibitory mechanism such as that affecting ocular motor preparatory activity, but a more general difficulty in deploying inhibitory control, perhaps originating from dysfunction at some other neural site such as prefrontal cortex (McDowell et al. 2002). Studies have shown that inhibition is not a unitary phenomenon but has several components at different behavioral levels (De Jong et al. 1990, 1995). In the generation of an antisaccade, volitional control processes may be required to trigger the specific inhibitory processes that operate during a successful antisaccade. Difficulties might occur because of impairments in those control processes rather than in specific ocular motor inhibitory mechanisms: in this sense, antisaccade errors may reflect a failure to implement inhibition, rather than a deficit in inhibitory mechanisms. (An analogy may be that failure of a car to stop may not reflect a failure of the brakes, but a failure of the driver to engage the brake pedal.) If so, and the increased antisaccade error rates originate in impaired inhibitory control outside of the frontal eye field and superior colliculus, then the longer latencies of correct antisaccades could be explained as an adaptive attempt of intact inhibitory mechanisms to reduce ocular motor preparatory activity, to counteract a higher-level failure of implementation of inhibition.
In addition to implementing inhibition, antisaccades require the generation of the novel response of looking in the opposite direction (Munoz and Everling 2004). Therefore another possibility is that the primary failure lies in the generation of the novel antisaccade response. If the neural computations involved in generating an antisaccade take longer in schizophrenia subjects, then the greater time elapsed between target onset and antisaccade initiation could increase the probability that an automatic prosaccade would escape before that antisaccade could be initiated.
To distinguish between a generalized problem with implementing inhibition versus a specific problem with novel response generation, it would be useful to examine these processes separately. In the present study, we examined whether the implementation of inhibition independent of the requirement to generate a novel response is impaired in schizophrenia, by studying the act of simply maintaining fixation. The command to keep one’s gaze steadily on an immobile target requires the suppression of extraneous prosaccades to other stimuli in the environment. This may be more demanding in the context of a saccade experiment, when one is often required to maintain steady fixation on a starting point yet be prepared to make a saccade once the target appears. However, maintaining fixation does not involve any of the vector-inversion computations (i.e., inverting stimulus location into saccadic direction) involved in antisaccade generation. Therefore failures to maintain fixation are attributable to a defect affecting the implementation of inhibitory control, rather than a defect in the generation of novel responses.
In this report healthy controls and schizophrenia subjects performed a pseudorandom series of fixation, prosaccade and antisaccade trials. (This differs from the majority of antisaccade studies in schizophrenia, which have subjects perform prosaccades and antisaccades in separate blocks.) As with the majority of eye movement experiments, saccade trials also required that participants maintained a steady fixation on a central point and made a saccade only when the stimulus appeared. We hypothesized that an inconsistent implementation of inhibitory control of saccades would generate not only increased antisaccade error rates but also a higher rate of fixation losses in schizophrenia, both during the fixation trials and in the pre-target phase of saccade trials. Furthermore, if problems with inhibitory control contribute significantly to antisaccade errors, then antisaccade errors and fixation losses should be correlated across subjects. On the other hand, if increased antisaccade error rates are due solely to problems with generating the novel antisaccade, then there would be no grounds to expect a correlation between antisaccade errors and fixation losses. Third, we hypothesized that a common factor of impaired inhibitory control in antisaccade errors and fixation losses would lead to more antisaccade errors occurring on trials with unstable fixation than on trials with stable fixation.
Last, as a final investigation into the nature of the problems with inhibitory control in subjects with high rates of antisaccade errors, we asked whether prosaccade errors and antisaccade errors are also correlated across subjects. A common view of antisaccade errors is that they are due to the difficulty in inhibiting the relatively automatic response that is a prosaccade (Levy et al. 1998). However, when making an error on a prosaccade trial, the subject actually fails to execute the more automatic response, and instead makes an antisaccade. Hence the common view that antisaccade errors reflect a primary failure to inhibit prosaccades would not predict that errors on antisaccade trial and errors on prosaccade trials should be correlated.
Methods
Participants
Table 1 provides demographic information. The schizophrenia sample consisted of 15 chronic outpatients recruited from an urban community mental health center, who had been maintained on stable doses of a variety of atypical antipsychotic medications for at least 6 weeks. Diagnoses of schizophrenia were confirmed with Structured Clinical Interviews for DSM-IV (First et al. 1997). Clinical status was characterized with the Brief Psychiatric Rating Scale (Overall and Gorham 1962) and with the Positive and Negative Syndrome Scale (Kay et al. 1987).
Sixteen healthy control participants, without a personal history of psychiatric illness or a family history of schizophrenia spectrum disorders, were recruited from the community with poster advertisements. All subjects were screened to exclude substance abuse in the preceding 6 months or neurological conditions that might impair cerebral function. The control group was matched to the subject group for gender, parental education and socioeconomic status (Hollingshead 1965), and handedness as determined by the modified Edinburgh Handedness Inventory (scores of 70 and above denote strong right-hand preference) (White and Ashton 1976). Controls had more years of education and higher estimated premorbid verbal IQs as measured by a test of single word reading (Blair and Spreen 1989). We did not match the groups for education or IQ since schizophrenia interferes with educational attainment and is associated with reduced IQ, which predates the onset of illness, and then further declines (Seidman et al. 2006). Thus, fewer years of education and lower IQ may reflect cognitive deficits that are core to schizophrenic pathology, and not potential artifacts that should be controlled (Meehl 1970). All participants gave written informed consent. The study was approved by institutional review boards at Massachusetts General Hospital and the Massachusetts Department of Mental Health.
Procedure
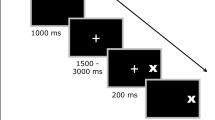
We used electrooculography to record eye movements during a magnetoencephalographic recording session. Signals were recorded continuously at 600 samples/s and minimally filtered (bandwidth of 0.1–200 Hz); the system has a sensitivity of at least 1°. The subject’s head was placed inside a helmet-shaped lower end of a Dewar containing the sensors, as they sat in an armchair facing a screen. The saccadic task stimuli were generated using the Vision Shell programming platform (http://www.visionshell.com) and presented with a digital light processing (DLP) InFocus 350 projector, through an opening in the wall, onto a back-projection screen placed 102 cm in front of the participant inside the magnetically shielded room. The system was calibrated by having the subject make prosaccades to 10-degree targets before starting the experimental blocks. The experimental paradigm consisted of a pseudorandom sequence of prosaccade, antisaccade, and fixation trials. The saccadic trials were balanced for right and leftward movements and lasted 4,000 ms (Fig. 1). Trials began with a 300-ms instructional cue at screen center. For half of the participants, an orange ring and blue cross were the cues for prosaccade and antisaccade trials, respectively. The cues were reversed for the remaining participants. The cue was flanked horizontally by two small green squares of 0.2° that marked the potential locations of stimuli, 10° left and right of center. These dots remained on the screen for the duration of each run. The cue was then replaced by a white fixation ring at the center with a diameter of 0.4° and a luminance of 20 cd/m2. After 1,700 ms, the ring shifted to one of the two stimulus locations chosen at random. This ring was the stimulus to which the participant responded. The ring remained in the peripheral location for 1,000 ms and then returned to the center, where participants were required to return their gaze for 1,000 ms before the start of the next trial. Randomly interleaved with the saccadic trials were 2, 4, or 6 s fixation trials, during which participants were instructed to maintain steady gaze at the center of the same screen display that was at the end of each saccadic trail. Each participant performed eight runs of the task with short rest breaks between runs. Each run was of 5 min 22 s duration and consisted of a random sequence of 26–46 prosaccades, 24–46 antisaccades, and 10–17 fixation trials. The total experiment lasted approximately 1 h and generated a total of 278 prosaccade, 285 antisaccade, and 107 fixation trials.

Saccadic paradigm. Top two rows show eye and target horizontal position against time, while the bottom row shows examples of the displays. Trials began with an instructional cue at screen center: for half of the participants, an orange ring cued a prosaccade trial and a blue X cued an antisaccade trial. These cues were reversed for the other half. The cue was flanked by two small squares that marked potential stimulus locations 10° left and right of center, which remained on the screen for the duration of each run. At 300 ms the instructional cue was replaced by a fixation ring at screen center. After 1,700 ms the ring shifted to one of the two stimulus locations with equal probability. The ring remained in the peripheral location for 1,000 ms and then returned to the center, where participants were instructed to return their gaze
To mitigate any potential motivational deficits in our schizophrenia subjects, all participants were offered a monetary reward of $0.05 for each correct response. The subjects were told to respond as quickly and as accurately as possible, to fixate their gaze on the central fixation point when not fixating on the peripheral target, and not to blink until after they have made the eye-movement.
Analysis
EOG data were scored in MATLAB (Mathworks, Natick MA) using a partially automated program that determined the directional accuracy of each saccade with respect to the required response and the latency from target onset. Saccades were identified as horizontal eye movements with velocities exceeding 47 deg/s. The onset of a saccade was defined as the point at which the velocity of the eye first exceeded 31 deg/s. Only those epochs with saccades in the desired direction with latencies between 130 and 800 ms were included for further analysis. The cutoff of 130 ms excluded anticipatory saccades, which are not true responses to the appearance of the visual target (Fischer and Breitmeyer 1987; Doricchi et al. 1997; Straube et al. 1999). Trials with eye-blinks (defined as vertical peak-to-peak EOG amplitude exceeding 200 μV) prior to saccadic response were rejected from further analysis. On average 209 ± 43 prosaccade and 201 ± 59 antisaccade trials were eligible for analysis for each participant.
We then tabulated the fixation losses during saccade trials by identifying the number of saccades occurring during the period between cue and target onset, when subjects were required to maintain steady fixation on the central stimulus. We also calculated fixation losses during fixation trials. Antisaccade errors were identified as trials in which the first saccade was towards the target rather than in the direction opposite to the target.
For each subject we calculated the frequency of saccade trials with fixation losses, the frequency of fixation trials with fixation losses, and the frequency of antisaccade errors and prosaccade errors, defined as trials in which the first saccade after stimulus onset had a horizontal vector component in the direction opposite to that desired (no responses are also defined as errors). We first compared the frequency of trials with fixation losses among schizophrenia subjects with that in healthy controls, using ANOVA with group (control vs. schizophrenia) and trial type (fixation, antisaccade, prosaccade) as main factors, and subjects nested within group as a random effect. Next we performed a correlation analysis of antisaccade error rates with either the frequency of fixation losses in all saccadic trials or the frequency of fixation losses in fixation trials, testing the null hypothesis that antisaccade error frequency was not significantly related to the frequency of fixation losses. This was done for each of the two subject groups. To determine if the relationship between fixation loss frequency and antisaccade errors differed between the two groups, we performed a multiple linear regression with antisaccade error rate as the dependent variable and subject group and fixation loss frequency as the main factors. Third, we tested whether the frequency of antisaccade errors was higher in trials with fixation losses than in trials without fixation losses. Because the number of trials with fixation losses was small in many subjects, and the number of such trials with antisaccade errors even smaller, we did this as a group analysis using chi-square tests.
Last, we performed a correlation analysis of antisaccade error rates with prosaccade error rates, testing the null hypothesis that these were not significantly related to each other. Again, we performed a multiple linear regression with antisaccade error rate as the dependent variable and subject group and prosaccade error rate as the main factors.
Results
First, the number of fixation trials on which unwanted saccades occurred was greater in schizophrenia subjects than healthy controls (Table 2). The ANOVA showed a significant main effect of group (F(1, 30) = 4.78, P < 0.037); however, there was no significant main effect of trial type or interaction between group and trial type. (Since the linear contrast between prosaccade trials and antisaccade trials did not show any difference in fixation losses (t = 0.4, n.s.), we will combine these data in the rest of the report.) On prosaccade and antisaccade trials, schizophrenia subjects lost fixation on 4.35% (SD 3.65) of trials, compared to 1.11% (SD 1.27) of trials for controls. On fixation trials, schizophrenia subjects lost fixation on 4.95% (SD 4.40) of trials compared to 1.58% (SD 2.95) of trials for controls. To determine if fixation losses were due to fatigue and increasing inattention over the experimental period, we contrasted the frequency of fixation losses in the first half of the test session with that in the second half using t tests: no significant differences were found for either group for either fixation trials or saccade trials.
Second, there was a significant correlation between the frequency of fixation losses during saccade trials and antisaccade error rate (Fig. 2a) for both healthy controls (r = 0.38, F(1, 15) = 4.95, P = 0.03), and schizophrenia subjects (r = 0.45, F(1, 14) = 7.53, P = 0.01). In contrast to these findings, there was no correlation between the frequency of fixation losses on fixation trials and antisaccade error rate (Fig. 2b) either in healthy controls (r = −0.02, P = 0.94) or schizophrenia subjects (r = −0.02, P = 0.95).

Correlation analyses. a Each subject’s antisaccade error frequency is plotted as a function of their frequency of fixation losses in the interval leading up to target onset, showing a significant correlation. b Antisaccade error frequency is plotted as a function of fixation losses on fixation trials, which do not require preparation for a saccade. No correlation is found. c Antisaccade error frequency is plotted as a function of prosaccade error frequency, showing a significant correlation
Figure 2a suggests that one difference between controls and schizophrenia subjects is simply that the latter group has more subjects with high antisaccade error rates and high frequencies of fixation loss. However, one might also question whether the two groups differ in the slope of the relationship between antisaccade errors and fixation losses during saccade trials. To examine this, we performed a multiple linear regression analysis. This confirmed a significant effect of fixation loss frequency (F(1,31) = 4.81, P = 0.037), but no significant interaction between group and fixation loss frequency (F(1, 31) = 1.61, P = 0.22), indicating no difference in the effect of fixation loss frequency between the groups. In part this may be due to a single control outlier with high fixation loss but low antisaccade error rate: if the analysis is repeated omitting his data, there is a significant interaction between subject group and fixation loss frequency (F(1, 30) = 30.3, P < 0.001). Nevertheless, given the limited range of values for fixation loss frequency in the healthy controls, there is insufficient evidence to conclude that the relationship is either the same or different between the groups.
Third, we examined whether a fixation loss was a sign of instability in the saccadic system that would increase the likelihood of an antisaccade error in the same trial. We contrasted the antisaccade error rate on those trials on which fixation losses had occurred prior to the target’s appearance with the error rate on those antisaccade trials with steady fixation (Fig. 3). Including all subjects, trials with losses of fixations had a higher antisaccade error rate (38/185, or 20.5%) than trials with steady fixation (1,133/8,202, or 13.8%) (chi-square(df1) = 6.82, P = 0.009). This was also true for schizophrenia subjects alone (35/155, or 22.6%, vs. 636/3,845, or 16.5%) (chi-square (1) = 3.89, P = 0.049). (Control subjects alone did not have enough fixation loss trials with antisaccade errors for chi-square analysis.)

For each subject, the likelihood of antisaccade errors on trials with stable fixation is plotted against the likelihood of such errors on trials with unstable fixation. The diagonal line represents the points where these are equivalent. Overall, and particularly among the schizophrenia subjects, antisaccade errors are more likely after a period of unstable fixation
Last, we found (Fig. 2c) a significant positive correlation between prosaccade and antisaccade errors in both controls (r = 0.63, F(1, 15) = 18.91, P = 0.0002) and schizophrenia subjects (r = 0.46, F(1, 14) = 7.76, P = 0.009). Multiple linear regression analysis confirmed a significant main effect of prosaccade error rate (F(1, 31) = 13.03, P = 0.001), but no significant interaction between subject group and prosaccade error rate (F(1, 31) = 2.50, P = 0.13), indicating no difference in the effect of fixation loss frequency between the groups.
Discussion
This study produced four main findings. First, schizophrenia subjects are more prone to making unwanted saccades during fixation during both the preparatory interval for an upcoming saccade and also during periods of steady fixation interspersed among saccadic trials. Second, in both healthy and schizophrenia groups, subjects with a higher incidence of fixation losses during the preparatory period of a saccadic trial also had higher antisaccade error rates, but the incidence of fixation losses on fixation trials (when no saccade needed to be prepared) was not correlated with a subject’s antisaccade error rate. Third, on a trial by trial basis, a preparatory period in which fixation was interrupted by an unwanted saccade was more likely to be followed by an antisaccade error than a preparatory period when fixation had been successfully maintained. Thus, these findings provide indirect evidence for a link between antisaccade errors and the ability to suppress unwanted saccades during fixation, and support the assertion that failures in the implementation of inhibitory control contribute significantly to impaired antisaccade performance in schizophrenia. Our last finding, that antisaccade errors were significantly correlated with prosaccade errors, challenges the conventional assertion that this inhibitory control is specifically concerned with suppressing the more automatic prosaccade response.
Previous studies on fixation behavior in schizophrenia have yielded mixed results. A number of studies have reported normal fixation stability (Clementz et al. 1994; Radant et al. 1997; Kissler and Clementz 1998; Gooding et al. 2000; Hutton et al. 2002), while others find deficits (Mialet and Pichot 1981; Matsue et al. 1986; Amador et al. 1991; Paus 1991; Curtis et al. 2001; Raemaekers et al. 2002). These studies varied not only in their results but also in the nature of their fixation tasks, however, and a review of task parameters suggests important differences that may explain some of the variability in results. Most of the studies with normal results examined fixation on a stationary spot without distractors and for periods of 5–30 s (Matsue et al. 1986; Ross et al. 1988; Clementz et al. 1994; Kissler and Clementz 1998; Gooding et al. 2000). Studies reporting abnormal fixation tended to have differences in protocol, such as longer fixation periods of 45 s (Mialet and Pichot 1981), a requirement for a saccade between two targets to start the fixation period (Amador et al. 1991), or the interspersing of fixation epochs among saccades made every 5 s (Curtis et al. 2001). Increased fixation instability in schizophrenia has also be seen in conditions such as darkness or with eyes closed (Matsue et al. 1986). Most consistently, though, problems with fixation losses in schizophrenia have been manifest on trials in which distractors intermittently appeared in the peripheral field while the subject tried to maintain fixation on a central target (Paus 1991; Curtis et al. 2001; Raemaekers et al. 2002) (but see Hutton et al. 2002). Such tasks are actually “no-go” tasks, in that they require subjects to inhibit the natural tendency to make a saccade towards a suddenly appearing target (Barton et al. 2006).
Our study shows additional conditions that may lead to fixation instability in schizophrenia subjects. We too assessed fixation epochs interspersed with saccade trials, but also assessed fixation behavior just prior to an impending saccade. We hypothesize that the recent execution of saccades or the imminent need to execute a saccade may place the saccadic system in a primed or activated state, making it easier for fixation instability to be manifest than in paradigms in which fixation is measured in isolation from any saccadic task. This priming of the saccadic system would also be suggested by our finding of a greater likelihood of an antisaccade error following a fixation loss in the pre-target period. It would also explain why abnormal fixation stability was more likely to be found in prior studies that assessed fixation after a saccade, interspersed among saccades, or with distractors that may have served as potential saccadic targets.
The correlation of fixation instability with antisaccade errors provides support for the hypothesis that difficulties implementing inhibition are an important factor in the generation of the high-antisaccade error rate in schizophrenia. While some studies have found a correlation between fixation and smooth pursuit abnormalities in schizophrenia (Mialet and Pichot 1981; Matsue et al. 1986; Amador et al. 1991, 1995), there has been little prior investigation of the relationship between fixation and antisaccade performance. One study (Curtis et al. 2001) found a correlation between the frequency of fixation losses and the antisaccade error rate, particularly between a no-go fixation task and antisaccades with an overlap condition, in which the central fixation spot persists after appearance of the peripheral target, making it harder to disengage fixation. They argued that adding distractors to the fixation task and adding the overlap design to the antisaccade task increased the inhibitory load of the two tasks, and that the resulting stronger correlation between fixation and antisaccade performance would imply a defect in inhibitory control, consistent with our hypothesis.
In our study, although fixation losses were increased in schizophrenia in both fixation and saccade trials, only those during saccade trials were correlated with antisaccade error rate across the whole group. Thus the likelihood of maintaining fixation during trials when saccadic processes are being mobilized is predictive of the error rate when the saccade is actually executed. Furthermore, antisaccade trials in which fixation was lost were more likely to be followed by an error. Thus, on both a subject-by-subject basis and a trial-by-trial basis, loss of fixation during the preparation for a saccade is predictive of the likelihood of making an antisaccade error, further strengthening the relation between the inhibitory control required during fixation and that needed during antisaccade execution. Furthermore, our finding of a similar relationship between antisaccade error rate and fixation losses on saccadic trials in healthy subjects suggests that this link between inhibitory control during fixation and that during antisaccade execution is a general characteristic of human ocular motor performance.
Our final finding provided more information on the nature of this inhibitory control. If the failures to maintain steady fixation or to make antisaccades were due to an inability to suppress the nearly reflexive prosaccade response, then one would not expect a correlation with prosaccade errors, in which a subject fails to make a prosaccade rather than making an unwanted one. Since we did find a significant correlation, this suggests that the problem with implementation of inhibitory control does not lie specifically with suppressing prosaccades. Rather, a problem suppressing both unwanted prosaccades and unwanted antisaccades suggests a more general problem with implementing inhibition of erroneous responses. Others have suggested that this difficulty in implementing inhibition may originate in deficits in attentional maintenance, working memory (Roberts et al. 1994; Nieman et al. 2000; Gooding and Tallent 2001), or goal activation (Nieuwenhuis et al. 2004), any of which could be consistent with our findings (but see Donohoe et al. (2006) for a contrary view on the contribution of working memory vs. inhibition). Deficits in these processes may also explain the finding that higher antisaccade error rates and longer antisaccade latencies are correlated in some studies with impaired smooth pursuit in schizophrenia (Sereno and Holzman 1995). Furthermore, such higher-level deficits can account for correlations of antisaccade deficits with impairments on the Wisconsin card sort test (Rosse et al. 1993; Crawford et al. 1995, 1996), the trail making test (Radant et al. 1997; Nieman et al. 2000) and with tests of spatial working memory and the continuous performance test, a probe of maintained vigilance (Nieman et al. 2000).
Mention should also be made of the potential role of medications in our study, which examined only treated schizophrenia subjects. As such, our report cannot provide any direct data on medication effects. However, two studies have confirmed that increased antisaccade error rates are found in neuroleptic-free schizophrenia subjects (Crawford et al. 1995; Müller et al. 1999), and other studies have noted a lack of correlation between antisaccade error rate and neuroleptic dose (Fukushima et al. 1990a; Rosse et al. 1993; Maruff et al. 1998; Raemaekers et al. 2002). For fixation stability, two studies found no effect of type of medication (atypical vs. typical antipsychotic vs. other drugs) on fixation losses (Gooding et al. 2000; Curtis et al. 2001), while two other studies reported no difference between medicated and unmedicated subjects (Amador et al. 1995; Kissler and Clementz 1998). Thus the available evidence on both fixation and antisaccade performance suggests that the findings we report are likely directly related to schizophrenia and are not by-products of the treatment of the condition.
References
Amador X, Sackeim H, Mukherjee S, Halperin R, Neeley P, Maclin E, Schnur D (1991) Specificity of smooth pursuit eye movement and visual fixation abnormalities in schizophrenia. Comparison to mania and normal controls. Schizophr Res 5:135–144
Amador X, Malaspina D, Sackeim H, Coleman E, Kaufman C, Hasan A, Brown J (1995) Visual fixation and smooth pursuit eye movement abnormalities in subjects with schizophrenia and their relatives. J Neuropsychiatr 7:197–206
Barton J, Raoof M, Jameel O, Manoach D (2006) Task-switching with antisaccades versus no-go trials: a comparison of inter-trial effects. Exp Brain Res 172:114–119
Blair J, Spreen O (1989) Predicting premorbid IQ: a revision of the national adult reading test. Clin Neuropsychol 3:129–136
Boudet C, Bocca M, Chabot B, Delamillieure P, Brazo P, Denise P, Dollfus S (2005) Are eye movement abnormalities indicators of genetic vulnerability to schizophrenia? Eur Psychiatry 20:339–345
Broerse A, Crawford T, den Boer J (2001) Parsing cognition in schizophrenia using saccadic eye movements: a selective overview. Neuropsychologia 39:742–756
Calkins M, Iacono W, Curtis C (2003) Smooth pursuit and antisaccade performance evidence trait stability in schizophrenia subjects and their relatives. Int J Psychophysiol 49:139–146
Calkins M, Curtis C, Iacono W, Grove W (2004) Antisaccade performance is impaired in medically and psychiatrically healthy biological relatives of schizophrenia subjects. Schizophr Res 71:167–178
Clementz B, McDowell J, Zisook S (1994) Saccadic system functioning among schizophrenia subjects and their first-degree biological relatives. J Abnorm Psychol 103:277–287
Crawford T, Haeger B, Kennard C, Reveley M, Henderson L (1995) Saccadic abnormalities in psychotic subjects. I. Neuroleptic-free psychotic subjects. Psychol Med 25:461–471
Crawford TJ, Puri BK, Nijran KS, Jones B, Kennard C, Lewis SW (1996) Abnormal saccadic distractibility in subjects with schizophrenia: a 99mTc-HMPAO SPET study. Psychol Med 26:265–277
Curtis C, Calkins M, Iacono W (2001) Saccadic disinhibition in schizophrenia subjects and their first-degree biological relatives. A parametric study of the effects of increasing inhibitory load. Exp Brain Res 137:228–236
De Jong R, Coles M, Logan G, Gratton G (1990) In search of the point of no return: the control of response processes. J Exp Psychol Hum Percept Perform 16:164–182
De Jong R, Coles M, Logan G (1995) Strategies and mechanisms in nonselective and selective inhibitory motor control. J Exp Psychol Hum Percept Perform 21:498–511
Donohoe G, Reilly R, Clarke S, Meredith S, Green B, Morris D, Gill M, Corvin A, Garavan H, Robertson I (2006) Do antisaccade deficits in schizophrenia provide evidence of a specific inhibitory function? J Int Neuropsychol Soc 12:901–906
Doricchi F, Perani D, Incoccia C, Grassi F, Cappa SF, Bettinardi V, Galati G, Pizzamiglio L, Fazio F (1997) Neural control of fast-regular saccades and antisaccade: an investigation using positron emission tomography. Exp Brain Res 116:50–62
Everling S, Munoz DP (2000) Neuronal correlates for preparatory set associated with pro-saccades and anti-saccades in the primate frontal eye field. J Neurosci 20:387–400
Everling S, Dorris MC, Klein RM, Munoz DP (1999) Role of primate superior colliculus in preparation and execution of anti-saccades and pro-saccades. J Neurosci 19:2740–2754
First M, Spitzer R, Gibbon M, Williams J (1997) Structured clinical interview for DSM-IV axis I disorders, research version, subject edition with psychotic screen (SCID-I/P W/PSY SCREEN). Biometrics Research, New York State Psychiatric Institute, New York
Fischer B, Breitmeyer B (1987) Mechanisms of visual attention revealed by saccadic eye movements. Neuropsychologia 25:73–83
Fukushima J, Morita N, Fukushima K, Chiba T, Tanaka S, Yamashita I (1990a) Voluntary control of saccadic eye movements in subjects with schizophrenic and affective disorders. J Psychiatr Res 24:9–24
Fukushima J, Fukushima K, Morita N, Yamashita I (1990b) Further analysis of the control of voluntary saccadic eye movements in schizophrenic subjects. Biol Psychiatry 28:943–958
Fukushima J, Fukushima K, Miyasaka K, Yamashita I (1994) Voluntary control of saccadic eye movement in subjects with frontal cortical lesions and parkinsonian subjects in comparison with that in schizophrenics. Biol Psychiatry 36:21–30
Gooding D, Tallent K (2001) The association between antisaccade task and working memory task performance in schizophrenia and bipolar disorder. J Nerv Ment Dis 189:8–16
Gooding D, Grabowski J, Hendershot C (2000) Fixation stability in schizophrenia, bipolar, and control subjects. Psychiatry Res 97:119–128
Gooding D, Mohapatra L, Shea H (2004) Temporal stability of saccadic task performance in schizophrenia and bipolar subjects. Psychol Med 34:921–932
Gottesman I, Gould T (2003) The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 160:636–645
Hallett P, Adams B (1980) The predictability of saccadic latency in a novel voluntary oculomotor task. Vis Res 20:329–339
Hollingshead A (1965) Two factor index of social position. Yale University Press, New Haven
Hutton S, Ettinger U (2006) The antisaccade task as a research tool in psychopathology: a critical review. Psychophysiology 43:302–313
Hutton S, Kennard C (1998) Oculomotor abnormalities in schizophrenia. A critical review. Neurology 50:604–609
Hutton S, Joyce E, Barnes T, Kennard C (2002) Saccadic distractibility in first-episode schizophrenia. Neuropsychologia 40:1729–1736
Kay S, Fiszbein A, Opler L (1987) The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull 13:261–276
Kissler J, Clementz B (1998) Fixation stability among schizophrenia subjects. Neuropsychobiology 38:57–62
Levy D, Mendell N, LaVancher C et al (1998) Disinhibition in antisaccade performance in schizophrenia. In: Lenzenweger M, Dworkin R (eds) Origins and development of schizophrenia. American Psychological Association, Washington DC, pp 185–210
Levy D, O’Driscoll G, Matthysse S, Cook S, Holzman P, Mendell N (2004) Antisaccade performance in biological relatives of schizophrenia subjects: a meta-analysis. Schizophr Res 71:113–125
Manoach DS, Lindgren KA, Cherkasova MV, Goff DC, Halpern EF, Intriligator J, Barton JJS (2002) Schizophrenic subjects show deficient inhibition but intact task-switching on saccadic tasks. Biol Psychiatry 51:816–825
Maruff P, Danckert J, Pantelis C, Currie J (1998) Saccadic and attentional abnormalities in subjects with schizophrenia. Psychol Med 28:1091–1100
Matsue Y, Okuma T, Saito H, Aneha S, Ueno T, Chiba H, Matsuoka H (1986) Saccadic eye movements in tracking, fixation, and rest in schizophrenic and normal subjects. Biol Psychiatry 21:382–389
McDowell J, Brown G, Paulus M, Martinez A, Stewart S, Dubowitz D, Braff D (2002) Neural correlates of refixation saccades and antisaccades in normal and schizophrenia subjects. Biol Psychiatry 51:216–223
Meehl P (1970) Nuisance variables and the ex post facto design. In: Radner M, Winokur S (eds) Minnesota studies in the philosophy of science. University of Minnesota Press, Minneapolis, pp 373–402
Mialet J, Pichot P (1981) Eye-tracking patterns in schizophrenia. Arch Gen Psychiatry 38:183–186
Müller N, Riedel M, Eggert T, Straube A (1999) Internally and externally guided voluntary saccades in unmedicated and medicated schizophrenic subjects. Part II. Saccadic latency, gain, and fixation suppression errors. Eur Arch Psychiatry Clin Neurosci 249:7–14
Munoz D, Everling S (2004) Look away: the anti-saccade task and the voluntary control of eye movement. Nat Rev Neurosci 5:218–228
Nieman D, Bour L, Linszen D, Goede J, Koelman J, Gersons B, Ongerboer de Visser B (2000) Neuropsychological and clinical correlates of antisaccade task performance in szchizophrenia. Neurology 54:866–871
Nieuwenhuis S, Broerse A, Nielen M, de Jong R (2004) A goal activation approach to the study of executive function: an application to antisaccade tasks. Brain Cogn 56:198–214
Overall J, Gorham D (1962) The brief psychiatric rating scale. Psychol Rep 10:799–812
Paus T (1991) Two modes of central gaze fixation maintenance and oculomotor distractibility in schizophrenics. Schizophr Res 5:145–152
Radant A, Claypoole K, Wingerson D, Cowley D, Roy-Byrne P (1997) Relationships between neuropsychological and oculomotor measures in schizophrenia subjects and normal controls. Biol Psychiatry 42:797–805
Radant A, Dobie D, Calkins M, Olincy A, Braff D, Cadenhead K, Freedman R, Green M, Greenwood T, Gur R, Light G, Meichle S, Mintz J, Nuechterlein K, Schork N, Seidman L, Siever L, Silverman J, Stone W, Swerdlow N, Tsuang M, Turetsky B, Tsuang D (2006) Successful multi-site measurement of antisaccade performance deficits in schizophrenia. Schizophr Res 89:320–329
Raemaekers M, Jansma J, Cahn W, Van der Geest J, van der Linden J, Kahn R, Ramsey N (2002) Neuronal substrate of the saccadic inhibition deficit in schizophrenia investigated with 3-dimensional event-related functional magnetic resonance imaging. Arch Gen Psychiatry 59:313–320
Reuter B, Kathmann N (2004) Using saccade tasks as a tool to analyze executive dysfunctions in schizophrenia. Acta Psychol (Amst) 115:255–269
Roberts R, Hager L, Heron C (1994) Prefrontal cognitive processes: working memory and inhibition in the antisaccade task. J Exp Psychol Gen 123:374–393
Ross D, Ochs A, Hill M, Goldberg S, Pandurangi A, Winfrey C (1988) Erratic eye tracking in schizophrenic subjects as revealed by high-resolution techniques. Biol Psychiatry 24:675–688
Rosse RB, Schwartz BL, Kim SY, Deutsch SI (1993) Correlation between antisaccades and Wisconsin card sorting test performance in schizophrenia. Am J Psychiatry 150:333–335
Seidman L, Buka S, Goldstein J, Tsuang M (2006) Intellectual decline in schizophrenia: evidence from a prospective birth cohort 28 year follow-up study. J Clin Exp Neuropsychol 28:225–242
Sereno A, Holzman P (1995) Antisaccades and smooth pursuit eye movements in schizophrenia. Biol Psychiatry 37:394–401
Straube A, Riedel M, Eggert T, Muller N (1999) Internally and externally guided voluntary saccades in unmedicated and medicated schizophrenic subjects. Part I. Saccadic velocity. Eur Arch Psychiatry Clin Neurosci 249:1–6
White K, Ashton R (1976) Handedness assessment inventory. Neuropsychologia 14:261–264
Acknowledgments
DSM was supported by the National Institute for Mental Health (R01 MH67720), National Center for Research Resources (P41-RR14075), and the Mental Illness and Neuroscience Discovery (MIND) Institute (DOE-DE-FG02-99ER62764). J. J. S. B. was supported by CIHR grant MOP-81270, a Canada Research Chair, and a Senior Scholar award from the Michael Smith Foundation for Health Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barton, J.J.S., Pandita, M., Thakkar, K. et al. The relation between antisaccade errors, fixation stability and prosaccade errors in schizophrenia. Exp Brain Res 186, 273–282 (2008). https://doi.org/10.1007/s00221-007-1235-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-007-1235-2