
Abstract
In Parkinson’s disease (PD), the striatal dopamine depletion and the following overactivation of the indirect pathway of the basal ganglia leads to very early disinhibition of the subthalamic nucleus (STN) that may contribute to the progression of PD by glutamatergic overstimulation of the dopaminergic neurons in the substantia nigra. Adenosine A2A antagonism has been demonstrated to attenuate the overactivity of the striatopallidal pathway. To investigate whether neuroprotection exerted by the A2A antagonist 8-(3-chlorostyryl)caffeine (CSC) correlates with a diminution of the striatopallidal pathway activity, we have examined the changes in the mRNA encoding for enkephalin, dynorphin, and adenosine A2A receptors by in situ hybridization induced by subacute systemic pretreatment with CSC in rats with striatal 6-hydroxydopamine(6-OHDA) administration. Animals received CSC for 7 days until 30 min before 6-OHDA intrastriatal administration. Vehicle-treated group received a solution of dimethyl sulfoxide. CSC pretreatment partially attenuated the decrease in nigral tyrosine hydroxylase immunoreactivity induced by 6-OHDA, whereas no modification of the increase in preproenkephalin mRNA expression in the dorsolateral striatum was observed. The neuroprotective effect of the adenosine A2A antagonist CSC in striatal 6-OHDA-lesioned rats does not result from a normalization of the increase in striatal PPE mRNA expression in the DL striatum, suggesting that other different mechanisms may be involved.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder that progresses over years affecting prominently the dopaminergic neurons of the substantia nigra pars compacta (SNc). Indeed, most of the disabling motor symptoms of PD are due to this neuronal loss and the concomitant dramatic reduction of the dopamine content in the striatum. Although several dopaminomimetic drugs are useful in relieving motor symptoms, none of them clearly diminishes or prevents the progression of the disease.
Recently, adenosine A2A receptor antagonists have appeared to have an anti-parkinsonian effect in several experimental models of PD (Kanda et al. 1998, 2000; Grondin et al. 1999; Koga et al. 2000; Pinna et al. 2001), and to reverse levodopa-induced motor fluctuations (Bové et al. 2002). Adenosine A2A receptors are mainly expressed in the striatum (Jarvis and Williams 1989; Ongini and Fredholm 1996; Moreau and Huber 1999; Svenningsson et al. 1999; Kaelin-Lang et al. 2000; El Yacoubi et al. 2001) and colocalized with preproenkephalin mRNA (Schiffmann et al. 1991; Augood and Emson 1994; Augood 1999) and dopamine (DA) D-2 receptor mRNA (Fink et al. 1992; Pollack et al. 1993; Johansson et al. 1997) in the striatopallidal medium spiny neurons that constitute the so-called indirect pathway of the basal ganglia. In PD (Miller and DeLong 1987; Bergman et al. 1990) and in experimental models (Mitchell et al. 1989; DeLong 1990), it has been demonstrated that this output pathway is overactive. This phenomenon is revealed by up-regulation of enkephalin and its encoding mRNA in humans (Grafe et al. 1985; Nisbet et al. 1995; Calon et al. 2002), monkeys (Asselin et al. 1994; Herrero et al. 1995; Jolkkonen et al. 1995; Morissete et al. 1997) and rats (Voorn et al. 1987; Gerfen et al. 1990; Jian et al. 1990; Engber et al. 1991; Nisenbaum et al. 1994; Carta et al. 2002). A deficient level of striatal dopamine and the following overactivation of the indirect pathway leads to a very early (Vila et al. 2000) disinhibition of the subthalamic nucleus (STN) and, subsequently, to excessive subthalamopallidal drive. This results in decreased facilitation of cortical motor areas and consequent development of akinesia and bradykinesia (Bergman et al. 1990).
In addition to its main targets, the STN also sends excitatory projections to the dopaminergic neurons in the SNc (Kita and Kitai 1987). Therefore, it has been postulated that the subthalamic desinhibition may also contribute to the progression of PD by glutamatergic overstimulation of SNc neurons, leading to a vicious circle in which STN overactivity and nigral damage support each other (Rodriguez et al. 1998). In agreement with this notion, it has been shown that reducing STN activity by means of local infusion of the glutamate antagonist MK801 (Blandini et al. 2001) or STN lesion (Piallat et al. 1996, 1999) protects SNc neurons from 6-OHDA neurotoxicity.
Adenosine A2A antagonism has been demonstrated to attenuate the overactivity of the striatopallidal pathway since systemic administration of adenosine A2A receptor antagonists reverses increased gammaminobutyric acid (GABA) release in the globus pallidus (Ochi et al. 2000) and reverses the increased expression of preproenkephalin (PPE) in the striatum of unilateral 6-OHDA-lesioned rats (Aoyama et al. 2002). On the basis of these data, it seems reasonable that adenosine A2A antagonists might exert a neuroprotective effect, at least in part, by counteracting striatopallidal pathway overactivity, and therefore reducing the glutamatergic input to the SNc from the STN.
With regard to neuroprotective activity, the A2A antagonists have shown to protect against neuronal damage in excitotoxicity (Jones et al. 1998a, b) and ischemic models (Von Lubitz et al. 1995; Bona et al. 1997; Monopoli et al. 1998). Recent experimental data, has also indicated that A2A antagonists have neuroprotective properties in PD models, specifically in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice and 6-OHDA-treated rats (Chen et al. 2001, 2002; Ikeda et al. 2002; Schwarzschild et al. 2002). In vitro studies have pointed out some possible mechanisms of its neuroprotective effect. On one hand, the A2A antagonist KW-6002 modifies the packaging of [3H]MPP+ into synaptic vesicles (Ikeda et al. 2002) and in the other hand, another A2A antagonist, 8-(3-chlorostyryl)caffeine (CSC), inhibits monoamine oxidase-B (MAO-B) (Chen et al. 2002), although it is still not clear how A2A antagonists exert their neuroprotective effect in PD experimental models.
The aim of the present study was to investigate whether neuroprotection exerted by CSC administration correlates with a diminution of the striatopallidal pathway activity. For this purpose, we examined the striatal changes in the mRNA encoding for enkephalin, dynorphin, and adenosine A2A receptors induced by subacute pretreatment with a selective A2A antagonist CSC (Moreau and Huber 1999) in an experimental model of PD in rats with striatal 6-OHDA administration.
Materials and methods
Animals and protocol treatments
Male Sprague-Dawley rats weighting 240–280 g and housed on a 12-h light/dark cycle with free access to food and water were used for the experiments. Animals received subacute administration of the selective A2A antagonist of 8-(3-chlorostyryl)caffeine (CSC, 5 mg/kg/day, ip, distributed in two injections, n=7; Sigma-Aldrich Co., Spain) for 7 days until 30 min before 6-OHDA intrastriatal administration. Vehicle-treated group received a solution of 2% DMSO ip (n=9). The dose of CSC used in the present study has been shown to potentiate levodopa effects in several behavioral paradigms (Bové et al. 2002). Animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the local Government.
Striatal 6-OHDA lesion
Animals were anaesthetized with sodium pentobarbital (50 mg/kg, ip) and placed in a stereotaxic frame with the incisor bar positioned at 0 for all injections. Unilateral stereotaxic injections of 6-OHDA (Sigma-Aldrich) were made into the left striatum using a Hamilton syringe. A concentration of 3.0 μg/μl of 6-OHDA hydrobromide dissolved in vehicle was injected into four striatal sites (2 μl/site, total dose 24 μg, n=16) at the following coordinates: (1) A: +1.4, L: +2.6, V: −5.0; (2) A: +0.4, L: +3.0, V: −5.0; (3) A: −0.4, L: +4.2, V: −5.0; (4) A: −1.3; L: +4.5, V: −5.0. These coordinates were calculated from bregma and according to the atlas of Paxinos and Watson (1982). Rate of injection was 1 μl/min, leaving the needle in place for a further 2 min before withdrawal. Rats were kept housed as before the experiment for 21 days allowing the progressive degeneration of the nigrostriatal system (Przedborski et al. 1995; Kirik et al. 1998). Sham-lesioned group received intrastriatal administration of 0.2% ascorbic acid/saline (n=5).

Time-course of experiments described in the text
Rotational behavior test
Rotational behavior induced by 0.5 mg/kg, sc apomorphine (Sigma-Aldrich) administration was measured 21 days after striatal 6-OHDA microinjections. Rats were placed in circular cages and tethered to an automated rotometer. The number of complete (360°) turns made during each 5-min period was recorded by computer. Rats were allowed 15 min to habituate to the rotometer before the administration of apomorphine. The total time of testing after apomorphine administration was of 1 h. Total rotational activity was measured by counting the total number of net contralateral turns after the deduction of the ipsilateral rotations.
Tissue collection
The day after apomorphine administration, rats were killed with an overdose of anesthesia. Brains were quickly removed from the skull and then frozen on dry ice and kept at −80°C until were cut on a cryostat (Leica, Spain). Coronal 14-μm thick sections were collected through the striatum and the substantia nigra pars compacta onto APTS (3-amino-propyltriethoxysilane; Sigma-Aldrich) coated slides, and kept at −40°C until used.
Tyrosine hydroxylase immunohistochemistry
Nigral and striatal sections were defrozed and dried at room temperature and fixed with acetone for 10 min at 4°C. Then were rinsed in phosphate buffered saline (PBS) pH 7.4; Sigma-Aldrich) twice, 5 min each, and immersed in 0.3% hydrogen peroxide (Merck-Schuchardt, Hohennbrunn, Germany) in PBS for 10 min to block the endogenous peroxidase. At this point, sections were rinsed again in PBS and incubated with horse serum (GibcoBRL, Life Technologies Ltd, Auckland, New Zealand) with 0.1% Triton X-100 (Sigma-Aldrich) for 20 min. Sections were incubated overnight at 4°C with mouse anti-tyrosine hydroxylase (TH) monoclonal antibody (Chemicon Int. Inc., Calif., USA) at a dilution 1:500 in PBS. Sections were rinsed twice in PBS, 5 min each, and ImmunoPure Ultra-Sensitive ABC Peroxidase staining kit (Pierce, Ill., USA) was used to carry out the ABC staining method. By so doing, sections were incubated with biotinylated horse anti-mouse Ig-G for 30 min, followed by two rinses in PBS, and then incubated with avidin-biotinylated peroxidase complex for 30 min more. Finally, sections were rinsed in PBS and incubated with 3-3′-diaminobenzidine (Sigma-Aldrich) and 0.01% hydrogen peroxide for 15 min. Slides were washed with PBS, dehydrated in ascending alcohol concentrations, cleared in xylene and coverslipped in DPX-EXLI mounting medium.
TH-inmunoreactive (TH-IR) cell bodies were counted (10×, brightfield) in three consecutive sections per animal. The counting started at the first section where SNc was clearly separated from the ventral tegmental area by the medial terminal nucleus of the accessory optic tract. The optical densities of the TH-IR fibers in the striatum were measured in three slices per animal of the rostral level of the striatum, corresponding to the area around the second 6-OHDA injection. Sections were placed under a microscope connected via a video camera to a computer. Quantitative image analysis were performed with MCID computerized image analysis system (St Catherines, Ontario, Canada). The measured values (optical densities) were averaged for each rat and then expressed as relative percent from intact striatum of control animals.
In situ hybridization histochemistry
The oligonucleotides used were complementary to the following base sequences (GeneBank accession number in brackets): rat preprodynorphin, bases 607–654 [NM_019374]; rat preprodynorphin, bases 489–533 [NM_019374]; rat preproenkephalin, bases 513–542 [K02807]; human adenosine A2A receptor, bases 285–329 [NM_00675]. They were custom-synthesized by Amersham Pharmacia Biotech (UK). The oligonucleotides were labeled at their 3′-end by using [α−33P]dATP (Amersham, UK) and terminal deoxynucleotidyl-transferase (Roche Molecular Biochemicals, Mannheim, Germany). Labeled probes were purified trough QIAquick Nucleotide Removal columns (Qiagen, Germany).
For in situ hybridization, frozen tissue striatal sections were brought to room temperature, air-dried, and fixed for 20 min in 4% paraformaldehyde in phosphate-buffered saline (1× PBS: 2.6 mM KCl, 1.4 mM KH2 PO4, 136 mM NaCl, and 8 mM Na2 HPO4), washed once in 3×PBS, twice in 1×PBS, 5 min each, and incubated in a freshly prepared solution of predigrested pronase (Calbiochem, San Diego, Calif., USA) at a final concentration of 20 IU/ml in 50 mM TrisHCl pH 7.5, 5 mM EDTA for 2 min at 20°C. Proteolytic activity was stopped by inmersion for 30 s in 2 mg/ml glycine in PBS. Tissues were rinsed in PBS and dehydrated in graded ethanol 2 min each. For hybridization, labeled probes were diluted to a final concentration of 107 cpm/ml in a solution containing 50% formamide, 4×SSC (1×SCC: 150 mM NaCl, 15 mM sodium citrate), 1×Denhardt’s solution (0.02% Ficoll, 0.02% polyvinylpyrrolidone, 0.02% bovine serum albumin), 1% sarkosyl, 10% dextran sulphate, 20 mM phosphate buffer, pH 7.0, 250 μg/ml yeast tRNA, and 500 μg/ml salmon sperm DNA. Tissues were covered with 100 μl of the hybridization solution and overlaid with Nescofilm (Bando Chemical, Kobe, Japan) coverslips to prevent evaporation. Sections were incubated in humid boxes overnight at 42°C and then washed 4 times (45 min each) in 600 mM NaCl, 20 mM TrisHCl, pH 7.5, 1 mM EDTA at 60°C. Hybridized sections were exposed to BIOMAX-MR film (Kodak) for 15 days depending on the probe used at −70°C with intensifying screens.
The specificity of the nucleotide hybridization signals was assessed as follows. For a given oligonucleotide probe the presence of a 50-fold excess of the same unlabeled oligonucleotide in the hybridization buffer resulted in the abolishment of the specific hybridization signal (data not shown). The thermal stability of the hybrids was examined by washing a series of consecutive hybridized sections at increasing temperatures. Specific hybridization signals were still present in sections washed at 70°C but they were completely absent from sections washed at 80°C. No such decrease was observed in the background levels of the signal (data not shown).
The striatum were divided into two portions for the mRNA expression measurement, including the dorsolateral and the ventromedial striatum (Carta et al. 2002). Quantitative image analysis were performed with MCID computerized image analysis system (St Catherines).
Statistical analysis
Data were analyzed by analysis of variance (ANOVA) followed by Dunnett’s t-test for multiple comparisons. The level of statistical significance was set at P<0.05 for all analysis.
Results
Substantia nigra cell counts
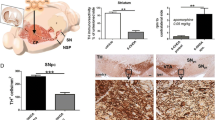
We assessed the effect of the adenosine A2A receptor antagonist CSC on the 6-OHDA-induced dopaminergic neuronal degeneration in rats. In this experiment, CSC or vehicle were subacutely (7 days) administered before striatal 6-OHDA lesion. Striatal 6-OHDA administration induced a decrease in the number of TH-IR neurons in the ipsilateral SNc of the vehicle-treated group by 48% in comparison with contralateral SNc (P<0.01). Sham-lesioned animals did not show any difference between both sides. Interestingly, CSC subacute pretreatment conferred a significant attenuation of the 6-OHDA-induced decrease in the number of TH-IR neurons by about 12% (P<0.05) (Fig. 1).
Striatal TH-immunoreactivity
In the vehicle-treated group, TH-IR decreased after striatal 6-OHDA administration by 20% in the ipsilateral striatum in comparison to contralateral striatum (P<0.01), while in the sham-lesioned animals no changes in the TH-IR was observed. Subacute CSC pretreatments failed to attenuate the decrease in the TH-IR induced by striatal 6-OHDA administration (P<0.05) (Fig. 2).

Effect of unilateral striatal 6-OHDA-induced lesion and CSC pretreatment on nigral TH-IR. Upper: a representative TH immunohistochemistry. Bottom: subacute administration of the A2A antagonist CSC partially attenuated the decrease in TH-IR when administered for 7 days until 30 min before 6-OHDA administration. Vehicle-treated group received a solution of 2% DMSO IP Sham-lesioned group received intrastriatal administration of 0.2% ascorbic acid/saline. Values are expressed as mean±SEM.**P<0.01 vs intact side. #P<0.05 vs vehicle-treated animals
Rotational behavior
Apomorphine induced rotational behavior to the 6-OHDA-lesioned vehicle-treated animals (P<0.01). Sham-lesioned animals did not show rotational behavior after apomorphine administration. Subacute CSC pretreatment did not modify the rotational behavior achieved by the 6-OHDA lesioned animals (Table 1).
Striatal adenosine A2A receptor mRNA expression
Levels of the different mRNA were measured in the dorsolateral (DL) and ventromedial (VM) portion of the striatum (Fig. 3). Striatal 6-OHDA administration induced a significant increase of A2A receptor mRNA levels in the VM, but not in the DL lesioned striatum, compared with the sham-lesioned animals (P<0.05). Subacute CSC pretreatment prevented this increase of A2A receptor mRNA levels induced by the striatal 6-OHDA administration (P<0.05) (Figs. 4 and 5).

Effect of unilateral striatal 6-OHDA-induced lesion and CSC pretreatment on striatal TH-IR. Subacute administration of the A2A antagonist CSC did not attenuate the decrease in TH-IR when administered for 7 days until 30 min before 6-OHDA administration. Vehicle-treated group received a solution of 2% DMSO IP Sham-lesioned group received intrastriatal administration of 0.2% ascorbic acid/saline. Values are expressed as mean±SEM. *P<0.05, **P<0.01 vs intact side

Schematic representation of striatal portions considered to measure mRNA expression by in situ hybridization

Effect of unilateral striatal 6-OHDA-induced lesion and CSC pre-treatment on DL (upper) and VM (bottom) striatal adenosine A2A mRNA expression. Subacute administration of the A2A antagonist CSC attenuated the increase in A2A mRNA expression in the VM striatum induced by intrastriatal 6-OHDA lesion. Values are expressed as mean±SEM. *P<0.05 vs sham-lesioned animals, #P<0.05 vs vehicle-treated animals
Striatal preproenkephalin mRNA expression
A significant increase in PPE mRNA levels in the DL lesioned striatum was caused by the 6-OHDA administration in the vehicle-treated animals compared with the sham-lesioned animals (P<0.01). In the VM striatum, 6-OHDA administration did not induce any change on PPE mRNA levels. Subacute CSC treated animals did not modify the increase in PPE mRNA expression induced by 6-OHDA in the DL striatum (Figs 6, 7).

Representative film autoradiograms of coronal brain sections (14 μm) showing striatal A2A mRNA labeling in control (sham-lesioned), vehicle-treated and CSC-treated rats

Effect of unilateral striatal 6-OHDA-induced lesion and CSC pretreatment on DL (upper) and VM (bottom) striatal PPE mRNA expression. Subacute administration of the A2A antagonist CSC did not attenuate the increase in PPE mRNA expression in the DL striatum induced by intrastriatal 6-OHDA lesion. Values are expressed as mean±SEM. **P<0.01 vs sham-lesioned animals
Striatal preprodynorphin mRNA expression
Striatal 6-OHDA administration induced no changes in PPD mRNA levels neither in the DL or in the VM striatum in the vehicle-treated animals compared with the sham-lesioned animals. Subacute CSC pretreatment produced a significant decrease in mRNA PPD levels in the lesioned VM striatum compared with sham-lesioned animals (P<0.05) (Figs 8, 9, 10).

Representative film autoradiograms of coronal brain sections (14 μm) showing striatal PPE mRNA labeling in control (sham-lesioned), vehicle-treated and CSC-treated rats

Effect of unilateral striatal 6-OHDA-induced lesion and CSC pretreatment on DL (upper) and VM (bottom) striatal PPD mRNA expression. Subacute administration of the A2A antagonist CSC decreased PPD mRNA expression in the VM striatum. Values are expressed as mean±SEM. *P<0.05 vs sham-lesioned animals; #P<0.01 versus vehicle-treated animals

Representative film autoradiograms of coronal brain sections (14 μm) showing striatal PPD mRNA labeling in control (sham-lesioned), vehicle-treated and CSC-treated rats
Discussion
In the present study, a four-site terminal lesion resulted in a partial loss of TH-positive fibers in the striatum, leading to a retrograde degeneration of the 48% of dopaminergic neurons in the SNc. Systemic CSC administration partially attenuated nigral dopaminergic cell loss induced by intrastriatal 6-OHDA administration. These results are in agreement with previous reports that demonstrated a neuroprotective effect of A2A antagonists in excitoxicity (Jones et al. 1998a,b; Behan and Stone 2002) and in ischemia models (Von Lubitz et al. 1995; Bona et al. 1997; Monopoli et al. 1998; Melani et al. 2003). Moreover, it has been recently described a neuroprotective effect of A2A receptor blockade in experimental models of PD since it has been shown that caffeine and selective A2A antagonists such as CSC, but not A1 antagonists, attenuated MPTP toxicity in mice (Chen et al. 2001, 2002; Xu et al. 2002). In 6-OHDA-treated rats, the selective A2A antagonist KW-6002 has shown to protect against both the loss of nigral dopaminergic cells and the degeneration of its terminals (Ikeda et al. 2002). In the present study, CSC administration did not attenuate the decrease in striatal TH-IR induced by intrastriatal 6-OHDA indicating a lack of protection of striatal dopaminergic terminals. This result agrees with the observation that the rotational behavioral showed by the group of animals pretreated with CSC did not differ from the vehicle-treated animals. Two methodological differences need to be taking in account to interpret the different results in comparison to a previous report (Ikeda et al. 2002). First of all, a much higher total dose of 6-OHDA has been used in the present study and it has been injected at four different sites of the striatum and not at a single one. In fact, a four-site 6-OHDA lesion has been compared with a manifest symptomatic stage in PD, whereas one-site 6-OHDA injection causes more restricted presymptomatic lesions (Kirik et al. 1998). The second methodological difference is the treatment protocol used since in the present study CSC was subacutely administered for 7 days until 30 min before 6-OHDA lesion. However, in the work of Ikeda et al. (2002), the A2A antagonist, KW-6002 was administered before the 6-OHDA administration and during 1 week later.
The precise mechanisms underlying the neuroprotective effect of A2A antagonists are still not known. Since there are evidences of the existence of functional A2A receptor in nigral dopaminergic neurons, it is possible that these neurons might be the site of the neuroprotective action by A2A antagonists (Okada et al. 1996; Chen et al. 2000). However, different A2A receptor-mediated mechanisms may be involved in central actions of A2A antagonists. For example, A2A receptor stimulation enhances striatal glutamate extracellular levels (Simpson et al. 1992; Popoli et al. 1995; Sebastiao and Ribeiro 1996) and the A2A antagonist SCH 58261 decreases both spontaneous and K+-evoked striatal glutamate outflow in rats (Corsi et al. 2000). Since glutamate is considered to play a major role inducing ischemia and post-ischemia cell death (Choi and Rothman 1990), protective effects of A2A -receptor antagonists against ischemic injury may be attributed to their ability to reduce excitatory amino acid outflow.
Several previous studies have involved A2A receptors in cerebral inflammation (Sullivan et al. 1999) and therefore adenosine might contribute to the pathological changes in PD by triggering the activation of surrounding glial cells, which are known to appear around degenerating dopaminergic neurons in PD (Hirsch et al. 1999) since A2A receptor-mediated mechanisms have been described in substantia nigra (Alfinito et al. 2003). Although A2A receptors inhibit the production of several pro-inflammatory cytokines (Dianzani et al. 1994), they can also potentiate the pro-inflammatory effect of those compounds (Scholz-Pedretti et al. 2001). Activation of A2A receptors can promote glial proliferation after brain injury (Hindley et al. 1994; Rathbone et al. 1999) and enhances nitric oxide and cyclooxygenase production in vitro (Fiebich et al. 1996). However, another report suggests that adenosine may inhibit astroglial activation (Michael et al. 1999). The protective effect of A2A receptor antagonists may therefore reflect a net attenuation of pro-inflammatory activity.
CSC is also a potent and selective inhibitor of monoamine oxidase-B (MAO-B) (Chen et al. 2002) and it has been suggested that the neuroprotective effect of this drug may be due to a blockade of the conversion of MPTP to MPDP+, an oxidation mediated by MAO-B, in the MPTP model of PD (Chen et al. 2002). The generation of reactive oxygen species induced by 6-OHDA may arise from two distinct mechanisms, namely deamination by MAO oxidation or auto-oxidation (Blum et al. 2001). Thus, 6-OHDA, like DA, may be a substrate for MAO (Breese and Taylor 1971; Karoum et al. 1993). An involvement of MAO in 6-OHDA-induced neurotoxicity has been suggested following the observation that the MAO inhibitor, selegiline, prevents 6-OHDA toxicity (Salonen et al. 1996) and, consequently, the inhibition of MAO by CSC could be one explanation for the CSC neuroprotective effects.
The restricted expression of A2A receptors in the striatum and the lack of evidence for their expression on dopaminergic neurons themselves (Rosin et al. 1998; Svenningsson et al. 1999) suggest that A2A receptors modulation of dopaminergic neurotoxicity is indirect either by an alteration in their retrograde neurotrophic influence in nigrostriatal neurons (Siegel and Chauhan 2000) or more likely through a feedback circuit running back to the dopaminergic nigral neurons (Rodriguez et al. 1998). In the latter case, stimulation of A2A receptors on striatopallidal neurons enhances GABA release in the globus pallidus (Mayfield et al. 1996) and thus may facilitate the indirect pathway disinhibition of STN activity, which in turn through the glutamatergic projections to the SNc may contribute to excitotoxic injury of dopaminergic neurons (Piallat et al. 1996). Inactivation of A2A receptors, on the other hand, would prevent the proposed dopaminergic toxicity produced through this circuit.
In order to investigate the possible involvement of the indirect and the direct striatopallidal pathways activity changes in the neuroprotection induced by CSC administration we have study the expression of striatal mRNA expression for adenosine A2A receptor, PPE and PPD in rats with a striatal 6-OHDA-induced lesion. We have shown that 6-OHDA intrastriatal administration produce a significant increase in adenosine A2A receptor mRNA expression in the VM striatum, but not in the DL, ipsilateral to the lesion. These results are in agreement with a recent report (Pinna et al. 2002) in which the expression of adenosine A2A receptor mRNA was increased in the striatum in association with a decrease in striatal extracellular levels of adenosine. The increase was selectively detected in the lateral portion of the lesioned striatum which partially overlaps the portion that in the present study has been defined as VM striatum. As has been proposed (Pinna et al. 2002), the specific distribution of A2A receptors to the lateral portion of the striatum may account for the lack of changes in A2A mRNA expression when the whole striatum was studied (Kaeling-Lang et al. 2000). Binding studies have failed to demonstrate a modification of A2A receptor after 6-OHDA-induced denervation (Alexander and Reddington 1989; Martinez-Mir et al. 1991; Morelli et al. 1994; Przedbordki et al. 1995). These discrepancies between receptor binding and hybridization have been attributed to different sensitivities of the two methodologies (Pinna et al. 2002). In the present study, CSC pretreatment prevented the A2A receptor mRNA up-regulation in the VM striatum. This result suggests that the neuroprotective effect of CSC might be induced by an attenuation of the increased activity of the indirect pathway in which neuronal A2A receptors are expressed.
With the objective to investigate whether the attenuation of the hyperactivity of the indirect pathway is involved in the neuroprotective effect of A2A antagonism we have studied the expression of PPE mRNA, since its increase has been correlated to the hyperactivity of this pathway (Young et al. 1986; Gerfen et al. 1990; Cadet et al. 1992; Asselin et al. 1994). We have shown that striatal 6-OHDA administration increased the PPE mRNA levels in the DL lesioned striatum in agreement with previous descriptions after striatal (Winkler et al. 2002) and after nigrostriatal lesions induced by 6-OHDA (Young et al. 1986; Gerfen et al. 1990; Cadet et al. 1992; Zeng et al. 1995) or MPTP administration (Augood et al. 1989; Asselin et al. 1994; Jolkkonen et al. 1995). The most relevant finding in the present study is that CSC pretreatment did not attenuate this increase in PPE mRNA in the DL lesioned striatum. Since the increase in PPE mRNA may reflect an overactivity of the striatopallidal indirect pathway leasing to increased inhibition of pallidal neurons and subsequent overactivity of STN (Levy et al. 1997; Parent et al. 2000), the results obtained in the present work suggest that the neuroprotective effect of A2A antagonist CSC is not related to an attenuation of the indirect striatopallidal pathway.
In the present study, no modification of PPD mRNA levels has been induced by intrastriatal 6-OHDA lesion in agreement with the level of denervation of the lesioned striatum as previously showed (Winkler et al. 2002). CSC pretreatment induced a decrease in the expression of dynorphin mRNA in the VM striatum in rats with a striatal 6-OHDA-induced lesion. The role of this decrease in the expression of PPD in the VM lesioned striatum is not known. The VM striatum appears to play a critical role in mediating motoric effects (Boye et al. 2001; Ikemoto 2002; Ikemoto and Witkin 2003). It has been suggested that the A2A receptors, localized in the ventral striatum play a key role in the modulation of motor activity. Barraco et al. (1993) showed that the local infusion in the VM of the selective A2A agonist CGS21680, but not a selective A1, induced a pronounced motor depressant in mice. As far as the VM striatum is concerned, low doses of caffeine stimulate spontaneous motor activity (Svenningsson et al. 1995). Morphological observations suggest that GABAergic striopallidal neurons and strionigral-strioentopeduncular neurons might be the main locus for A2A-D2 and A1-D1 interactions, respectively (Schiffmann et al. 1991; Fink et al. 1992). The two subtypes of GABAergic efferent neurons are also present in the VM striatum (LeMoine and Bloch 1995), although with a less well-defined separation of their target brain areas. Although A2A and D1 receptor are not located on the same striatal efferent neurons, there are several studies that clearly illustrate an A2A receptor modulation of the striatonigral pathway at behavioral and biochemical level in 6-OHDA-lesioned rats (Morelli et al. 1994; Pinna et al. 1996; Pollack and Fink 1996). It has been shown that systemic administration of the A2A antagonist SCH 58261 caused a decrease in the number of c-fos mRNA-containing neurons in the striatum not only in the striatopallidal pathway but in the striatonigral pathway (Le Moine et al. 1997). A2A receptor antagonism-induced potentiation of D1 receptor-mediated motor activation has been demonstrated (Pinna et al. 1996). All these effects could be explained by an interaction at the network level, similar to the synergistic effect of dopamine D1 and D2 agonists (Robertson and Robertson 1986; Paul et al. 1992).
Since synaptic connections between spiny neurons of the direct and indirect pathways have been described (Aronin et al. 1986; Yung et al. 1996; Seeman and Tallerico 2003), A2A antagonists could modulate the direct pathway via the indirect pathway. The existence of such functional interaction between adenosine A2A receptors and dopamine D1 receptors may underlie the effect of the administration of CSC diminishing PPD mRNA expression in the VM striatum shown in the present study. Furthermore, the increase of dynorphin mRNA levels seen after chronic levodopa treatment in 6-OHDA lesioned mice is not seen in A2A knockout mice (Freduzzi et al. 2002), demonstrating that A2A receptors are involved in dynorphin mRNA levels modulation and therefore in striatonigral pathway activity. These results are in agreement with our results showing that A2A blockade attenuates dynorphin expression. The role of this decrease in PPD mRNA expression in the VM striatum on the neuroprotective effect of CSC is not known. However, a cytotoxic effect of dynorphin has been described (McIntosh et al. 1994; Hauser et al. 1999; Tan-No et al. 2001). Thus, it might be speculated that a decrease in dynorphin might have a neuroprotective effect.
In summary, the present results show that the neuroprotective effect of the adenosine A2A antagonist CSC in striatal 6-OHDA-lesioned rats does not result from a normalization of the increase in striatal PPE mRNA expression in the DL striatum suggesting that other different mechanisms may be involved. A recent hypothesis of a different role of A2A receptors at pre- versus postsynaptic sites on neuroprotection needs to be taken in account, since it has been shown (Tebano et al. 2004) that whereas effects of presynaptic A2A receptors are potentially detrimental, the effects of postsynaptic A2A receptors are potentially beneficial.
References
Alexander SP, Reddington M (1989) The cellular localization of adenosine receptors in rat neostriatum. Neuroscience 28:645–651
Alfinito PD, Wang SP, Manzino L, Rijhsinghani S, Zeevalk GP, Sonsolla PK (2003) Adenosinergic protection of dopaminergic and GABAergic neurons against inhibition through receptors located in the substantia nigra and striatum. J Neurosci 23:10982–10987
Aoyama S, Koga K, Mori A, Miyaji H, Sekine S, Kase H, Uchimura T, Kobayashi H, Kuwana Y (2002) Distribution of adenosine A2A receptor antagonist KW6002 and its effect on gene expression in the rat brain. Brain Res 953:119–125
Aronin N, DiFiglia M, Graveland GA, Schwartz WJ, Wu JY (1984) Localization of immunoreactive enkephalin in GABA synthesizing neurons of the rat neostriatum. Brain Res 300:376–380
Aronin N, Chase K, DiFiglia M (1986) Glutamic acid decarboxylase and enkephalin immunoreactive axon terminals in the rat neostriatum synpase with striatonigral. Brain Res 365:151–158
Asselin MC, Soghomonian JJ, Cote PY, Parent A (1994) Striatal changes in preproenkephalin messenger RNA levels in parkinsonian monkeys. Neuroreport 5:2137–2140
Augood SJ (1999) Localization of adenosine A2A receptors in brain: therapeutic implications. Adv Neurol 80:105–109
Augood SJ, Emson PC, Mitchell IJ, Boyce S, Clarke CE, Crossman AR (1989) Cellular localisation of enkephalin gene expression in MPTP-treated cynomolgus monkeys. Brain Res Mol Brain Res 6:85–92
Augood SJ, Emson PC (1994) Adenosine A2A receptor mRNA is expressed by enkephalin cells but not somatostatin cells in rat striatum:a co-expression study. Mol Brain Res 22:104–210
Barraco RA, Martens KA, Parizon M, Normile HJ (1993) Adenosine A2A receptors in the nucleus accumbens mediate locomotor depression. Brain Res Bull 31:397–404
Behan WMH, Stone TW (2002) Enhanced neuronal damage by co-administration of quinolinic acid and free radicals, and protection by adenosine A2A receptor antagonists. Br J Pharmacol 135:1435–1442
Bergman H, Wichmann T, DeLong MR (1990) Reversal of experimental parkinsonism by lesion of the subthalamic nucleus. Science 249:1436–1438
Blandini F, Nappi G, Grenamyre JT (2001) Subthalamic infusion of an NMDA antagonist prevents basal ganglia metabolic changes and nigral degeneration in a rodent model of Parkinson’s disease. Ann Neurol 49:525–529
Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM (2001) Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol 65:135–172
Bona E, Aden U, Guilland E, Fredholm BB, Hagberg H (1997) Neonatal cerebral hypoxiaischemia: the effect of adenosine receptor antagonists. Neuropharmacology 9:1327–1338
Bové J, Marin C, Bonastre M, Tolosa E (2002) Adenosine A2A antagonism reverses levodopa-induced motor alterations in hemiparkinsonian rats. Synapse 46:251–257
Boye SM, Grant RJ, Clarke PBS (2001) Disruption of dopaminergic transmission in nucleus accumbens core inhibits the locomotor stimulant effects of nicotine and d-amphetamine in rats. Neuropharmacology 40:792–805
Breese GR, Taylor TD (1971) Depletion of brain noradrenaline and dopamine by 6-hydroxydopamine. Br J Pharmacol 42:88–99
Cadet JL, Zhu SM, Angulo JA (1992) Quantitative in situ hybridization evidence for differential regulation of proenkephalin and dopamine D2 receptor messenger RNA levels in rat striatum: effects of unilateral intrastriatal injections of 6-hydroxydopamine. Mol Brain Res 12:59–67
Calon F, Birdi S, Rajput AH, Hornykiewicz O, Bédard PJ (2002) Increase of preproenkephalin mRNA levels in the putamen of Parkinson’s disease patients with levodopa-induced dyskinesias. J Neuropathol Exp Neurol 61:186–196
Carta AR, Pinna A, Cauli O, Morelli M (2002) Differential regulation of GAD67, enkephalin and dynorphin mRNAs by chronic-intermittent L-dopa and A2A receptor blockade plus l-dopa in dopamine-denervated rats. Synapse 44:166–174
Chen JF, Beilstein M, Xy YH, Turner TJ, Moratalla R, Stundaert DG, Aloyo VJ, Fink JS, Schwarzschild MA (2000) Selective attenuation of psychostimulant-induced behavioral responses in mice lacking A2A adenosine receptors. Neuroscience 97:195–204
Chen JF, Xu K, Petzer JP, Staal R, Xu YH, Beilstein M, Sonsalla P, Castafnoli K, Castagnoli N Jr, Schwarzschild MA (2001) Neuroprotection by caffeine and A2A adenosine receptor inactivation in a model of Parkinson’s disease. J Neurosci 21:16
Chen JF, Steyn S, Staal R, Petzer JP, Xu K, Van der Schyf C, Castagnoli K, Sonsalla PK, Castagnoli N Jr, Schwarzschild MA (2002) 8-(3-Chlorostyryl)caffeine may attenuate MPTP neurotoxicity through dual actions of monoamine oxidase inhibition and A2A receptor antagonism. J Biol Chem 277:36040–36044
Choi DW, Rothman SM (1990) The role of glutamate neurotoxicity in hypoxic–ischemic neuronal death. Annu Rev Neurosci 13:171–182
Corsi C, Melani A, Bianchi L, Pedata F (2000) Striatal A2A adenosine receptor antagonism differentially modifies striatal glutamate outflow in vivo in young and aged rats. Neuroreport 11:2591–2592
De Long MR (1990) Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13:281–285
Dianzani C, Brunelleschi S, Viano I, Fantozzi R (1994) Adenosine modulation of primed human neutrophils. Eur J Pharmacol 263:223–226
El Yacoubi M, Ledent C, Parmentier M, Ongini E, Costentin J, Vaugeois JM (2001) In vivo labeling of the adenosine A2A receptor in mouse brain using the selective antagonist [3H]SCH58261. Eur J Neurosci 14:1567–1570
Engber TM, Susel Z, Kuo S, Ge CR, Chase TN (1991) Levodopa replacement therapy alters enzyme activities in striatum and neuroppetide content in striatal output regions of 6-hydroxydopamine lesioned rats. Brain Res 552:113–118
Fiebich BL, Biber K, Lieb K, van Calker D, Berger M, Bauer J, Gebike-Haerter PJ (1996) Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2A receptors. Glia 18:152–160
Fink JS, Weaver DR, Rivkees SA, Peterfreund RA, Pollack A, Adler EM, Reppert SM (1992) Molecular cloning of the rat A2A adenosine receptor. Selective co-expression with D-2 dopamine receptors in rat striatum. Mol Brain Res 14:186–195
Fredduzzi S, Moratalla R, Monopoli A, Cuellar B, Xu K, Ongini E, Impagnatiello F, Schwarzschild MA, Chen JF (2002) Persistent behavioral sensitization to chronic l-dopa requires A2A adenosine receptors. J Neurosci 22:1054–1062
Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Sibley DR (1990) D1 and D2 dopamine receptor regulated gene expression of striatonigral and striatopallidal neurons. Science 250:1429–1432
Grafe MR, Forno LS, Eng LF (1985) Immunocytochemical studies of substance P and Met-enkephalin in the basal ganglia and susbtantia nigra in Huntington’s, Parkinson’s and Alzheimer’s diseases. J Neuropathol Exp Neurol 44:47–59
Grondin R, Bédard PJ, Hadj Tahar A, Gregoire L, Mori A, Kase H (1999) Antiparkinsonian effect of a new selective adenosine A2A receptor antagonist in MPTP-treated monkeys. Neurology 52:1673–1677
Hauser KF, Foldes JK, Turbek CS (1999) Dynorphin A (1–13) neurotoxicity in vitro: opioid and non-opioid mechanisms in mouse spinal cord neurons. Exp Neurol 160:361–375
Herrero MT, Augood SJ, Hirsch EC, Javoy-Agid EC, Luquin MR, Agid Y, Obeso JA, Emson PC (1995) Effects of l-dopa on preproenkephalin and preprotachykinin gene expression in the MPTP-treated monkey striatum. Neuroscience 68:1189–1198
Hindley S, Herman MAR, Rathbone MP (1994) Stimulation of astrogliosis in vivo by extracellular ADP or an adenosine A2 receptor agonist. J Neurosci Res 38:399–406
Hirsch EC, Hunot S, Damier P, Brugg B, Faucheux BA, Michel PP, Ruberg M, Muriel MP, Mouatt-Prigent A, Agid Y (1999) Glial cell participation in the degeneration of dopaminergic neurons in Parkinson’s disease. Adv Neurol 80:9–18
Ikeda K, Kurokawa M, Aoyama S, Kuwana Y (2002) Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J Neurochem 80:262–270
Ikemoto S (2002) Ventral striatum anatomy of locomotor activity induced by cocaine, d-amphetamine, dopamine and D-1/D-2 agonists. Neuroscience 113:939–955
Ikemoto S, Witkin BM (2003) Locomotor inhibition induced by procaine injections into the nucleus accumbens core, but not the medial ventral striatum: implication for cocaine-induced locomotion. Synapse 47:117–122
Jarvis MF, Williams M (1989) Direct autoradiographic localization of adenosine A2A receptors in the rat brain using the A2A selective agonist 3H-CGS21680. Eur J Pharmacol 168:243–246
Jian HK, McGinty JF, Hong, JS (1990) Differential modulation of striatonigral dynorphin and enkephalin by dopamine receptor subtypes. Brain Res 507:57–64
Johansson B, Georgiev V, Fredholm BB (1997) Distribution and postnatal ontogeny of adenosine A2A receptors in rat brain: comparison with dopamine receptors. Neuroscience 80:1187–1207
Jolkkonen J, Jenner P, Marsden CD (1995) l-Dopa reverses altered gene expression of substance P but not enkephalin in the caudateputamen of common marmosets treated with MPTP. Mol Brain Res 32:297–307
Jones PA, Smith RA, Stone TW (1998a) Protection against hippocampal kainate excitotoxicity by intracerebral administration of an adenosine A2A receptor antagonist. Brain Res 800:328–335
Jones PA, Smith RA, Stone W (1998b) Protection against kainate-induced excitotoxicity by adenosine A2A receptor agonists and antagonists. Neuroscience 85:229–237
Kaelin-Lang A, Liniger P, Probst A, Lauterburg T, Burgunder JM (2000) Adenosine A2A receptor gene expression in the normal striatum and after 6-OH-dopamine lesion. J Neural Transm 107:851–859
Kanda T, Jackson MJ, Pearce RKB., Nakamura J, Kase H, Kuwana Y, Jenner P (1998) Adenosine A2A antagonist:a novel antiparkinsonian agent that does not provoke dyskinesia in parkinsonian monkeys. Ann Neurol 43:507–513
Kanda T, Jackson MJ, Smith LA, Pearce RKB, Nakamura J, Kase H, Kuwana Y, Jenner P (2000) Combined use of adenosine A2A antagonist, KW-6002 with l-dopa or with selective D-1 or D-2 dopamine agonists increases antiparkinsonian activity but not dyskinesia in MPTP-treated monkeys. Exp Neurol 162:321–327
Karoum F, Chrapusta SJ, Egan MF, Wyatt RJ (1993) Absence of 6-hydroxydopamine in the rat brain after treatment with stimulants and other dopaminergic agents: a mass fragmentographic study. J Neurochem 61:1369–1375
Kirik D, Rosenbland C, Björklund A (1998) Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp Neurol 152:259–277
Kita H, Kitai ST (1987) Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol 260:435–452
Koga K, Kurokawa M, Ochi M, Nakamura J, Kuwana Y (2000) Adenosine A2A receptor antagonist KF17837 and KW6002 potentiate rotation induced by dopaminergic drugs in hemiparkinsonian rats. Eur J Pharmacol 408:249–255
Le Moine C, Bloch B (1995) D-1 and D-2 dopamine receptor gene expression in the rat striatum: sensitive cRNA probes demonstrate prominent segregation of D-1 and D-2 mRNAs in distinct neuronal populations of the dorsal and ventral striatum. J Comp Neurol 355:418–426
Le Moine C, Svenningsson P, Fredholm BB, Bloch B (1997) Dopamine-adenosine interactions in the striatum and the globus pallidus: inhibition of striatopallidal neurons through either D-1 or A2A receptor enhances D-1 receptor-mediated effects on c-fos expression. J Neurosci 17:8038–8048
Levy R, Hazrati LN, Herrero MT, Vila M, Hassani OK, Mouroux M, Ruberg M, Asensi H, Agid Y, Feger J, Obeso JA, Parent A, Hirsch EC (1997) Re-evaluation of the functional anatomy of the basal ganglia in normal and parkinsonian states. Neuroscience 76:335–343
Martinez-Mir MI, Probst A, Palacios JM (1991) Adenosine A2A receptors:selective localization in the human basal ganglia and alterations with disease. Neuroscience 42:697–706
Mayfield RD, Larson G, Orona RA, Zahniser NR (1996) Opposing actions of adenosine A2A and dopamine D2 receptor activation on GABA release in the basal ganglia: evidence for and A2A /D2 receptor interaction in globus pallidus. Synapse 22:132–138
McIntosh TK, Fernyak S, Yamakami I, Faden AI (1994) Central and systemic kappa-opioid agonists exacerbate neurobehavioral response to brain injury in rats. Am J Physiol 267:R665–R672
Melani A, Pantoni L, Bordoni F, Gianfriddo M, Bianchi K, Vannucchi MG, Bertorelli R, Monopoli A, Pedata F (2003) The selective A2A receptor antagonist SCH58261 reduces striatal transmitter outflow, turning behavior and ischemic brain damage induced by permanent focal ischemia in the rat. Brain Res 959:243–250
Michael PP, Marien M, Ruberg M, Colpaert F, Agid Y (1999) Adenosine prevents the death of mesencephalic dopaminergic neurons by a mechanism that involves astrocytes. J Neurochem 72:2074–2082
Miller WC, DeLong MR (1987) Altered tonic activity of neurons in the globus pallidus and subthalamic nucleus in the primate MPTP model of parkinsonism. In: Carpenter MB, Jayaraman A (eds) The basal ganglia II. Plenum, New York, pp 415–427
Mitchell IJ, Clarke CE, Boyce S, Robertson RG, Peggs D, Sambrook MA, Crossman AR (1989) Neural mechanisms underlying parkinsonian symptoms based upon regional uptake of 2-deoxyglucose in monkeys exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience 32:123–226
Monopoli A, Lozza G, Forlani A, Mattaveli A, Ongini E (1998) Blockade of adenosine A2A receptors by SCH58261 results in neuroprotective effects in cerebral ischemia in rats. Neuroreport 9:3955–3959
Moreau JL, Huber G (1999) Central adenosine A2A receptors. An overview. Brain Res 31:65–82
Morelli M, Fenu S, Pinna A, Di Chiara G (1994) Adenosine A2A receptors interact negatively with dopamine D1 and D2 receptors in unilaterally 6-hydroxydopamine-lesioned rats. Eur J Pharmacol 251:21–25
Morissette M, Coulet M. Soghomonian JJ, Blanchet PJ, Calon F, Bédard PJ, DiPaolo T (1997) Preproenkephalin mRNA expression in the caudate–putamen of MPTP monkeys after chronic treatment with the D-2 agonist U91356A in continuous or intermittent mode of administration: comparison with l-dopa therapy. Mol Brain Res 49:55–62
Nisbet AP, Foster OJF, Kingsbury A, Eve DJ, Daniel SE, Marsden CD, Lees AJ (1995) Preproenkephalin and preprotachykinin messenger RNA expression in normal human basal ganglia and in Parkinson’s disease. Neuroscience 66:361–376
Nisenbaum LK, Kitai ST, Crowley WR, Gerfen CR (1994) Temporal dissociation between changes in striatal enkephalin and substance P messenger RNAs following striatal dopamine depletion. Neuroscience 60:927–937
Ochi M, Koga K, Kurokawa M, Kase H, Nakamura J, Kuwana Y (2000) Systemic administration of adenosine A2A receptor antagonist reverses increased GABA release in the globus pallidus of unilateral 6-hydroxydopamine-lesioned rats: a microdialysis study. Neuroscience 100:53–62
Okada M, Mizuno K, Kaneko S (1996) Adenosine A1 and A2 receptor modulate extracellular dopamine levels in rat striatum. Neurosci Lett 212:53–56
Ongini E, Fredholm BB (1996) Pharmacology of adenosine A2A receptors. Trends Pharmacol Sci 17:364–372
Parent A, Sato F, Wu Y, Gauthier J, Levesque M, Parent M (2000) Organization of the basal ganglia: the importance of axonal collateralization. Trends Neurosci 23:S20–S27
Paul ML, Graybiel AM, David JC, Robertson JA (1992) D1-like and D2-like dopamine receptors synergistically activate rotation and c-fos expression in the dopamine-depleted striatum in a rat model of Parkinson’s disease. J Neurosci 12:3729–3742
Paxinos G, Watson C (1982) The rat brain in stereotaxic coordinates. Academic, New York
Piallat B, Benazzouz A, Benabid AL (1996) Subthalamic nucleus lesion in rats prevents dopaminergic nigral neurons degeneration after striatal 6-OHDA injection: behavioural and immunohistochemical studies. Eur J Neurosci 8:1408–1414
Piallat B, Benazzouz A, Benabid AL (1999) Neuroprotective effect of chronic inactivation of the subthalamic nucleus in a rat model of Parkinson’s disease. J Neural Transm Suppl 55:71–77
Pinna A, DiChiara G, Wardas J, Morelli M (1996) Blockade of A2A adenosine receptors positively modulates turning behaviour and c-Fos expression induced by D-1 agonists in dopamine denervated rats. Eur J Neurosci 8:1176–1181
Pinna A, Fenu S, Morelli M (2001) Motor stimulant effects of the adenosine A2A receptor antagonist SCH58261 do not develop tolerance after repeated treatments in 6-hydroxydopamine-lesioned rats. Synapse 39:233–238
Pinna A, Corsi C, Carta AR, Valentini V, Pedata F, Morelli M (2002) Modification of adenosine extracellular levels and adenosine A2A receptor mRNA by dopamine denervation. Eur J Pharmacol 446:75–82
Pollack AE, Fink JS (1996) Synergistic interaction between an adenosine antagonist and a dopamine D-1 agonist on rotational behaviour and striatal c-fos induction in 6-hydroxydopamine-lesioned rats. Brain Res 743:124–130
Pollack AE, Harrison MB, Wooten FG, Fink SJ (1993) Differential localization of A2A adenosine receptor mRNA with D-1 and D-2 dopamine receptor mRNA in striatal output pathways following a selective lesion of striatonigral neurons. Brain Res 631:161–166
Popoli P, Betto P, Reggio R, Ricciarello G (1995) Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur J Pharmacol 287:215–217
Przedborski S, Levivier M, Jiang H, Ferreira M, Jackson-Lewis V, Donaldson D (1995) Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neuroscience 67:631–647
Rathbone MP, Middlemiss PJ, Gysbers JW, Andrews C, Herman MAR, Reed JK, Ciccarelli R, Di Iorio P, Caciagli F (1999) Trophic effects of purines in neurons and glial cells. Prog Neurobiol 59:663–690
Robertson GS, Robertson HA (1986) Synergistic effects of a D1 and D2 dopamine agonists on turning behaviour in rats. Brain Res 384:387–390
Rodriguez MC, Obeso JA, Olanow CW (1998) Subthalamic nucleus-mediated excitotoxicity in Parkinson’s disease: a target for neuroprotection. Ann Neurol 44:S175–S188
Rosin DL, Robeva A, Woodard RL, Guyenet PG, Linden J (1998) Immunohistochemical localization of adenosine A2A receptors in the rat central nervous system. J Comp Neurol 401:163–186
Salonen T, Haapalinna A, Heinonen E, Suhonen J, Hervonen A (1996) Monoamino oxidase inhibitor selegiline protects young and aged rat peripheral sympathetic neurons against 6-hydroxydopamine-induced neurotoxicity. Acta Neuropathol 91:466–474
Schiffmann SN, Jacobs O, Vanderhaeghen JJ (1991) Striatal restricted adenosine A2A receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridization histochemistry study. J Neurochem 57:1062–1067
Scholz-Pedretti K, Pfeilschifter J, Kaskin M (2001) Potentiation of cytokine induction of groups IIA phospholipase A2 in rat mesangial cells by ATP and adenosine via the A2A adenosine receptor. Br J Pharmacol 132:37–46
Schwarzschild MA, Chen JF, Ascherio A (2002) Caffeinated clues and the promise of adenosine A2A antagonists in PD. Neurology 58:1154–1160
Sebastiao AM, Ribeiro JA (1996) Adenosine A2 receptor-mediated excitatory actions of the nervous system. Prog Neurobiol 48:167–189
Seeman P, Tallerico T (2003) Link between dopamine D-1 and D-2 receptors in rat and human striatal tissues. Synapse 47:250–254
Siegel GJ, Chauhan NB (2000) Neurotrophic factors in Alzheimer’s and Parkinson’s disease brain. Brain Res Rev 33:199–227
Simpson RE, O’Regan MH, Perkins LM, Phillis JW (1992) Excitatory transmitter amino acid release from the ischaemic rat cerebral cortex: effects of adenosine receptor agonists and antagonists. J Neurochem 58:1683–1690
Sullivan GW, Linden J, Buster BL, Scheld WM (1999) Neutrophil A2A adenosine receptor inhibits inflammation in a rat model of meningitis: synergy with the type IV phosphodiesterase inhibitor, rolipram. J Infect Dis 180:1550–1560
Svenningsson P, Nomikos GG, Fredholm BB (1995) Biphasic changes in locomotor behavior and in expression of mRNA for NGFI-A and NGFI-B in rat striatum following acute caffeine administration. J Neurosci 5:7612–7624
Svenningsson P, Le Moine C, Fisone G, Fredholm BB (1999) Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog Neurobiol 59:355–396
Tan-No K, Cebers G, Yakovleva T, Goh BH, Gileva I, Reznikov K, Aguilar-Santelises M, Hauser KF, Terenius L, Bakalkin G (2001) Cytotoxic effects of dynorphins through nonopioid intracellular mechanisms. Exp Cell Res 269:54–63
Tebano MT, Pintor A, Frank C, Domenici MR, Martire A, Pepponi R, Potenza RL, Grieco R, Popoli P (2004) Adenosine A2A receptor blockade differentially influences excitotoxic mechanisms at pre- and postsynaptic sites in the rat striatum. J Neurosci Res 77:100–107
Vila M, Perier C, Feger J, Yelnik J, Faucheux B, Ruberg M, Raisman-Vozari R, Agid, Hirsch EC (2000) Evolution of changes in neuronal activity in the subthalamic nucleus of rats with unilateral lesion of the substantia nigra assessed by metabolic and electrophysiological measurements. Eur J Neurosci 12:337–344
Von Lubitz DK, Lin RC, Jacobson KA (1995) Cerebral ischemia in gerbils: effects of acute and chronic treatment with adenosine A2A receptor agonist and antagonist. Eur J Pharmacol 287:295–302
Voorn P, Roest G, Groenewegen HJ (1987) Increase of enkephalin and decrease of substance P immunoreactivity in the dorsal and ventral striatum of the rat midbrain 6-hydroxydopamine lesions. Brain Res 412:391–396
Winkler C, Kirik D, Björklund A, Cenci MA (2002) l-Dopa-induced dyskinesia in the striatal 6-hydroxydopamine model of Parkinson’s disease: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis 10:165–186
Xu K, Xu YH, Chen F, Schwarzschild MA (2002) Caffeine’s neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity shows no tolerance to chronic caffeine administration in mice. Neurosci Lett 322:13–16
Young WS, Bonner TI, Brann MR (1986) Mesencephalic dopamine neurons regulate the expression of neuropeptide messenger RNAs in the rat forebrain. Proc Natl Acad Sci USA 83:9827–9831
Yung KKL, Smith AD, Levey AI, Bolam JP (1996) Synaptic connections between spiny neurons of the direct and indirect pathways in the neostriatum of the rat: evidence from dopamine receptor and neuropeptide immunostaining. Eur J Neurosci 8:861–869
Zeng BY, Jolkkonen J, Jenner P, Marsden CD (1995) Chronic l-dopa treatment differentially regulates gene expression glutamate decarboxylase, preproenkephalin and preprotachykinin in striatum of 6-hydroxydopamine-lesioned rat. Neuroscience 66:19–28
Acknowledgements
This research was supported by grants from the Ministerio de Sanidad y Consumo (FIS 01/1499) and from the Ministerio de Ciencia y Tecnología (SAF2000–0212) of Spanish Government. J.B. and J.S. were supported by a grant from the Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS). The research groups are a part of the Spanish network for the research of neurological diseases (CIEN, Centro de Investigación de Enfermedades Neurológicas).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bové, J., Serrats, J., Mengod, G. et al. Neuroprotection induced by the adenosine A2A antagonist CSC in the 6-OHDA rat model of parkinsonism: effect on the activity of striatal output pathways. Exp Brain Res 165, 362–374 (2005). https://doi.org/10.1007/s00221-005-2302-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-005-2302-1