
Abstract
To reveal the characteristics and formation mechanism of proteins in the secondary precipitate of soy sauce (SPSS), proteins in the supernatant of soy sauce were used as control, and SPSS was prepared by centrifugation in combination with lyophilization. The proteins in SPSS were isolated and identified by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and matrix-assisted laser desorption ionization-time of flight/time of flight mass spectrometry. Acidic polypeptide A1a of soy glycinin G1 (32–35 kDa) and basic polypeptide B3 of soy glycinin G4 (23 kDa) were verified as the predominant proteins (95.2 %) in SPSS. The amino acid composition, average hydrophobicity (HΦ avg), and the secondary structure of the proteins in SPSS were investigated by high-performance liquid chromatography and Fourier transform infrared spectroscopy. Results revealed that SPSS contained not only proteins but also free amino acids; the significantly higher average hydrophobicity and much lower contents of β-sheet and random coil were the main characteristics and reasons for the formation of proteins in SPSS.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Soy sauce, which originated in China over 2,500 years ago, is an indispensable seasoning in China and many other Asian countries [1]. It is widely used to improve the aroma and taste of food. The unique aroma and umami taste make it more and more popular all over the world [2]. Moreover, soy sauce has been proved to contain various bioactive components and possess anticarcinogenic, antioxidation, antimicrobial, antiplatelet, and immunomodulating activities [3–5]. However, the secondary precipitate (Fig. 1) is a main quality problem obstructed the advancement of soy sauce industry [6]. Compared with the primary precipitate (present during soy sauce sterilization), SPSS appears during storage and marketing, and thus, it seriously affects the appearance quality of the final product and the buying intention of customers. Therefore, it is of great significance to solve the problem of SPSS.

The secondary precipitate of soy sauce
Currently, the preliminary work has verified that the main components of SPSS were proteins, NaCl, and carbohydrates; proteins were regarded as a key reason for the precipitate formation of soy sauce and other beverage [7–9]. Thus, elucidation of the characteristics and formation mechanism of the proteins in SPSS was prerequisite and vital to solve the problem of SPSS. To the best of our knowledge, a Japanese team has purified and identified the promoters, isomers, and homologs of l-glutamic acid 5-n-butyl ester, of precipitate formation in soy sauce, which could influence the precipitate formation of Japanese-type soy sauce [10, 11]. Additionally, the amino acid and chemical compositions of SPSS have been investigated by a Chinese team [7]. However, the in-depth information on the characteristics and formation mechanism of the proteins in SPSS is still unclear.
Therefore, the objective of this work is to prepare the proteins in SPSS and reveal their characteristics and formation mechanism by SDS-PAGE, MALDI-TOF/TOF MS, HPLC, and FTIR analysis. This will contribute to the systematical knowledge of SPSS and shed some light on its solving way.
Materials and methods
Materials
Soy sauce (prepared by defatted soy meal, wheat flour, and salt) was commercially available from a local supermarket (Vanguard, Jiangsu, China). Details were as follows: high-salt-diluted state fermentation; formaldehyde nitrogen content ≥0.4 g/100 mL; food additives (caramel pigment, glutamate sodium, sodium benzoate, xanthan gum); origin of the product, Beijing. Production date: October 2012–December 2012.
Other chemicals used in this work are of the highest commercial grade and obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).
Preparation of the secondary precipitate of soy sauce
Soy sauce was centrifuged at 5,000×g for 30 min; the secondary precipitate was carefully collected and lyophilized at −40 °C; the yield of SPSS was 0.75 ± 0.09 g/L (n = 3). The resulting secondary precipitate and its supernatant were sealed in polyethylene bottles and kept in a refrigerator (4 °C) for proximate and amino acid composition analysis.
The secondary precipitate and its supernatant were dialyzed against flowing deionized water for 24 h using dialysis tubings with cutoff molecular weights of 5 and 1 kDa to remove salt (Spectrum Laboratories Inc., California, USA). Then, 150 mL of the supernatant and 150 mL of 20 % TCA were mixed by a magnetic stirring meter for 1 h in an ice bath. The turbid liquid was centrifuged at 2,070×g for 20 min. The precipitates from the supernatant and the deionized secondary precipitate were washed with acetone (w/v = 1:5), and then, the turbid liquids were centrifuged at 2,070×g for 20 min to collect the precipitates, respectively. Finally, the precipitates were lyophilized for further SDS-PAGE, MALDI-TOF/TOF MS, and FTIR analysis, respectively.
Proximate analysis
Water contents were determined by drying samples in an oven at 105 °C to constant weight. Contents of reducing sugar and total sugar were determined by AOAC method using Fehling’s solution [12]. The total nitrogen contents of the samples were estimated by the micro-Kjeldahl method, in which the samples were digested with a known quantity of concentrated sulfuric acid in the Kjeltec digestion apparatus (2300 Analyzer Unit, Foss Tecator AB, Höganas, Sweden). The protein content was calculated as 6.25× nitrogen content. Formaldehyde nitrogen was measured by titration method [1]. NaCl content was determined by titration method with AgNO3 [12]. Ash content was determined by incinerating samples in a muffle furnace at 550 °C for 24 h [12].
Validation of the secondary precipitate proteins
One gram of the secondary precipitate and 1,000 mL of 15.6 % NaCl solution with different pH (pH 3.0–11.5, 0.5 pH/interval) were mixed by a magnetic stirrer for 1 h, respectively. Then, the mixtures were centrifuged at 5,000×g for 30 min, and the protein contents of the supernatants were determined by the Lowry method using bovine serum albumin as the standard [13].
SDS-PAGE analysis
Proteins from the SPSS and the supernatant were dissolved in aqueous solutions containing 8 M urea, 60 mM DTT, 50 mM Tris, and 1 % SDS, and the protein contents were adjusted to approximately 1 mg/mL, respectively. SDS-PAGE was performed on a vertical slab gel with a thickness of 1.0 mm (BIO-RAD Mini-PROTEAN® Tetra System, California, USA) using a discontinuous buffer. The ratio of acrylamide and bisacrylamide was 30:1. The concentrations of the stacking gel and the separating gel were 4 and 14 %, respectively. The samples were heated at 95 °C for 5 min prior to loading in the gel slots. For each sample, 15 μL was applied to each lane. After the electrophoresis, the gel was stained in 0.25 % Coomassie blue (R-250) in 50 % TCA and destained in 7 % acetic acid (methanol: acetic acid: water = 227:37:236 (v:v:v)) and then scanned by a JEDA scan instrument (JD-801, Jiangsu, China). The relative percentage of each band in abundance was determined by the densitometric scanning technique.
MALDI-TOF/TOF MS analysis
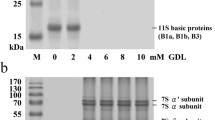
Each spot (a–h) in the SDS-PAGE gel (Fig. 2a–f) was cut out and subjected to in-gel trypsin digestion [14]. The peptide solution (1 μL) was applied to an Anchorchip™ target (Bruker-Daltonics, Bremen, Germany) with the same volume of matrix (10 mg/mL, α-Cyano-4-hydroxycinnamic acid in 50 % acetonitrile, 0.1 % trifluoroacetic acid) and analyzed on an Ultraflex™ MALDI-TOF/TOF MS (Bruker-Daltonics, Bremen, Germany). The tryptic peptides were analyzed in the positive ion reflector mode, and the spectra were calibrated using Bruker peptide calibration standard II (Bruker-Daltonics, Bremen, Germany). At least ten peptide fragments were analyzed in a lift mode. After spectra were processed by FlexAnalysis software and analyzed by BioTools software (Bruker-Daltonics, Bremen, Germany), an in-house Mascot server (http://www.matrixscience.com) was used for database search in the Green Plant or the Fungi taxonomy of NCBInr at the National Center for Biotechnology Information. The following criteria were used for the database search: tryptic digestion; monoisotopic peptide values; at least four matching peptide masses; a maximum of one missed cleavage per peptide; fragment mass tolerance of 0.5 Da and peptide mass tolerance of 100 ppm, together with the acceptance of cysteine carbamidomethylation (fixed modifications) and methionine oxidation (variable modifications). For a positive identification, a score calculated by the Mowse scoring algorithm in MASCOT was considered as significant (p < 0.05). The sequences encoding predicted proteins of unknown function were subjected to BLAST search in NCBI.

The representive electrophoresis map of proteins in SPSS and SSS. Lane M: standard markers. Lanes 1, 2 and 3, 4 indicate the electrophoresis profiles of SPSS and SSS, respectively. Alphabets a–h represent the pots (bands) were excised for MALDI-TOF/TOF MS identification. Bands a, b, c, and d (MW < 23.8 kDa) represent 11.4, 8.0, 75.8, and 4.8 % of the proteins in SPSS; bands e, f, g, and h (MW < 23.8 kDa) represent 0, 0, 7.0, and 93.0 % of the proteins in SSS
Amino acid composition analysis
The amino acid composition of samples was determined according to the method of Yang et al. [15] with a slight modification. Amino acid composition was determined by a high-performance liquid chromatography (Waters, Milford, Massachusetts, USA) equipped with a PICO. TAG column. Prior to amino acid composition analysis, all of the samples were filtered through micropore films (0.45 μm of pore size). Free amino acid composition was determined by injecting samples directly into chromatography system. The total amino acid composition of samples was determined after hydrolysis at 110 °C for 24 h with 6 M hydrochloric acid prior to the derivatization with phenyl isothiocyanate. External standards were used for quantification. The amino acid standards included l-alanine (Ala), l-arginine (Arg), l-aspartic acid (Asp), l-cystine (Cys), l-glutamic acid (Glu), l-glycine (Gly), l-histidine (His), l-isoleucine (Ile), l-leucine (Leu), l-lysine (Lys), l-methionine (Met), l-phenylalanine (Phe), l-proline (Pro), l-serine (Ser), l-threonine (Thr), l-tyrosine (Tyr), l-valine (Val), and ammonium chloride.
Average hydrophobicity analysis
In this work, content of individual amino acid in proteins was calculated as the difference value of its total content and that in free state. The average hydrophobicities of FAAs and proteins were analyzed according to the method of Lozano et al. [16]. The average hydrophobicities of the FAAs and the proteins in SPSS and SSS were calculated by the following equation:
where Xi is the molar ratio of the amino acid and HΦ i is the hydrophobicity of the amino acid.
FTIR analysis
Fourier transform infrared spectra of proteins in SPSS and SSS were recorded on a Nexus 470 spectrometer equipped with a deuterated triglycine sulfate (DTGS) detector and a single-reflection diamond attenuated total reflection (ATR) accessory (ThermoNicolet Nexus 470, Thermo Fisher Scientific, Waltham, Massachusetts, USA). Pellet containing 10 mg of sample and 200 mg of spectroscopy grade KBr was prepared using a Carver laboratory press with vacuum line attachment at 15,000 psi compression force. For each spectrum, 256 interferograms were collected with a resolution of 4 cm−1 with 32 scans and a 2 cm−1 interval from the 4,000 to 400 cm−1 region. Reference spectra were recorded under identical conditions but the medium (KBr) contained no protein. The subtraction of the reference spectrum from the spectrum of the samples was carried out in accordance with the criteria described below. The signal-to-noise ratio of these spectra (≫500) was high enough to perform the manipulation of subtraction. Deconvolution of infrared spectra was performed using the PeakFitv4.12 software (Systat Software Inc., San Jose, California, USA) in order to obtain the quantitative information of the secondary structure components of protein [17], and band assignment of proteins in the samples in the amide region (1,600–1,700 cm−1) was according to Byler et al. [18]. The areas of the assigned amide I bands in second-derivative spectra are linearly correlated with the amount of secondary structure in the proteins. According to this method, the secondary structure compositions of α-helix, β-sheet, β-turn, and random coil in proteins of the samples are listed in Table 3.
Statistical analysis
All determinations were conducted in triplicate, unless specified otherwise. The results were subjected to one-way ANOVA if necessary. Duncan’s new multiple range test was performed to determine the significant difference between samples within 95 % CI using SPSS 15.0 software (SPSS Inc., Chicago, Illinois, USA).
Results and discussion
Proximate analysis
Proximate analysis was conducted to understand the primary components of SPSS and SSS. As shown in Table 1, SPSS and SSS mainly comprised of proteins, NaCl, and carbohydrate (except for water). Proteins accounted for 26.63 % of the total weight in SPSS, indicating that proteins, the principal organic component, might be highly correlated with the formation of SPSS. The proteins content in SPSS was close to the result (25.97 %) of Zhang et al. [7], whereas significantly more abundant NaCl (41.17 %) and less abundant carbohydrate (7.40 %) were observed in this work. Different soy sauces and SPSS preparation methods might be responsible for the differences. Furthermore, formaldehyde nitrogen (1.41 %) and reducing sugar (0.87 %) in SPSS were also detected in this work, indicating that amino acids and (or) reducing sugar might be formed by their high hydrophobicities or/and the adsorption of large proteins and polysaccharides in SPSS.
Validation of the secondary precipitate proteins
To verify the hydrophobicity of the proteins in SPSS, the validation experiment was conducted. Results showed that there was no proteins detected in the 15.6 % NaCl solution, indicating that the proteins in SPSS could not dissolve in 15.6 % NaCl solutions with different pH (pH 3.0–11.5) and were the secondary precipitate proteins of soy sauce.
SDS-PAGE analysis of proteins in SPSS and SSS
The SDS-PAGE profiles of SPSS and SSS are shown in Fig. 2. Totally, 3 bands with molecular weights of about 35 kDa (band a), 32 kDa (band b), and 23 kDa (band c) were observed in SPSS, and only 1 slight band (23 kDa, band e) and a predominant blue zone (band h) with approximately molecular weight ranging from 1 to 17 kDa were observed in SSS. Previous works have confirmed that the bands with molecular weights about 35 and 20 kDa in the gel of SDS-PAGE were the acidic and basic polypeptides of soy glycinin, respectively [19, 20]. In addition, relative percentages of the predominant band c in SPSS and the predominant blue zone (band h) in SSS were 75.8 and 93.0 %, respectively, as estimated by the densitometric scanning technique (Data shown in the bottom of Fig. 2). As expected, SPSS mainly consisted of high molecular weight proteins, while SSS was mainly composed of low molecular weight proteins. The latter has been confirmed in our previous work [1]. Notably, protein with molecular weight of about 23 kDa (relative percentage 7.0 %) was also observed in SSS, indicating that the soluble protein (23 kDa) in supernatant might convert into the insoluble protein (23 kDa) of SPSS in strong ionic solution (soy sauce solution). Thus, it was reasonable to assume that the degraded fractions (23 kDa) of soy glycinin might gradually give rise to the formation of SPSS and constituted the major part of SPSS.
MALDI-TOF/TOF MS analysis of proteins in SPSS and SSS
It has been reported that the precipitate of soy sauce was mainly composed of proteins, which was caused by the complicated coagulation of proteins under the promoter action of isomers and homologs of l-glutamic acid 5-n-butyl ester [10]. However, the origin and detailed information of the proteins in precipitate are still unclear. To clarify the precise origin and detailed information of the proteins in SPSS, MALDI-TOF/TOF MS analysis was conducted. As shown in Fig. 2, a total of 8 spots selected from SDS-PAGE gel of SPSS (spots a–d) and SSS (spots e–h) were subjected to MALDI-TOF/TOF MS analysis. Results showed that 4 spots (a, b, c, g) were successfully identified as acidic polypeptide A1a of soy glycinin G1 (spots a, b) and basic polypeptide B3 of soy glycinin G4 (spots c, g), respectively, and the other 4 spots were unsuccessfully identified. Previous works pointed out that acidic polypeptide A1a of soy glycinin G1 and basic polypeptide B3 of soy glycinin G4 were polypeptides with molecular weights of about 37 kDa and 18–22 kDa, isoelectric points of 5.4 and 9.9, respectively [19–21]. In addition, Yuan et al. [19] and Mo et al. [22] proved that the solubilities of acidic polypeptide A1a of soy glycinin G1 and basic polypeptide B3 of soy glycinin G4 (especially the former) were much lower than those of other acidic polypeptides (i.e., A2, A3, A4) or glycinin, and the phenomenon was particularly remarkable when pH was below 5.0 (pH of soy sauce was 4.0–4.5). The result coincided with the fact that acidic polypeptide A1a of soy glycinin G1 and basic polypeptide B3 of soy glycinin G4 comprised of the major part of SPSS proteins and the latter accounted for about 75.8 % of the SPSS proteins. The much lower solubilities of acidic polypeptide A1a of soy glycinin G1 and basic polypeptide B3 of soy glycinin G4 (relative to other acidic polypeptides or glycinin) may be attributed to the presence of more hydrophobic amino acids and structure [19, 23]. Thus, it was necessary to investigate the characteristics of amino acid composition and the secondary structure of the proteins in SPSS and SSS.
Amino acid composition analysis of SPSS and SSS
Table 2 presents the amino acid compositions of SPSS and SSS. The contents of total amino acids and FAAs in SPSS were 20.43 and 7.01 %, respectively, which were significantly higher than those (3.62 and 2.64 %) in SSS. This was in agreement with the contents of protein and formaldehyde nitrogen in SPSS and SSS (Table 1). The amino acid compositions of SPSS and SSS were observed in Table 2, Glu was the amino acid with the highest content in SPSS and SSS, followed by Asp, while Cys was present with the lowest content in them. Similar amino acid compositions of SPSS, SSS, and soybean were found [24], which might be attributed to the main protein, soybean, used in soy sauce preparation. Furthermore, it was firstly confirmed a quite high level of FAAs (7.01 %) in SPSS. The result further proved the complexity of the N-containing compounds in SPSS. To further elucidate the characteristics and formation mechanism of proteins in SPSS, it was necessary to study their average hydrophobicity and higher structure.
Average hydrophobicities analysis of FAAs and proteins in SPSS and SSS
The average hydrophobicities( HΦ avg) of FAAs and proteins represented the formation propensity of SPSS and were measured by the method of Lozano et al. [16]. As shown in Fig. 3, the average hydrophobicities of FAAs and proteins in SPSS reached 1.08 and 0.77, which were 11.34 and 39.29 % higher than those in SSS, respectively. ANOVA showed that significant differences in the average hydrophobicities of FAAs and proteins were found between SPSS and SSS (p < 0.05). Zhang et al. [7] had deduced that SPSS might be formed under the hydrophobic interaction of the proteins in SPSS, especially under the condition of strong ionic solution (soy sauce solution). Report by Bigclow [25] demonstrated that high average hydrophobicity was a key reason for precipitate formation of protein. Furthermore, previous works showed that the solubilities of acidic and basic polypeptides of soy glycinin were positively correlated with their proportions of acidic amino acids (Glu and Asp) [19, 26]. In this case, the percentages of acidic amino acids (Glu and Asp) in proteins of SPSS and SSS reached 34.6 and 48.0 %, respectively, but the latter was 38.7 % higher than the former, suggesting that proteins in SPSS possessed a higher hydrophobicity, which was also in accordance with the fact that SPSS proteins were from precipitate and SSS proteins were from the supernatant. Thus, high average hydrophobicities (HΦ avg) of FAAs and proteins played a key role on the formation of SPSS.

The average hydrophobicities of FAAs and proteins in SPSS and SSS. Values in the twin columns followed by different letters are significantly different (p < 0.05)
FTIR analysis of proteins in SPSS and SSS
The secondary structure of protein is closely correlated with its hydrophobicity, and the latter shows negative correlation with its solubility [27]. Thus, investigation on the secondary structure of proteins in SPSS was propitious to elucidate their formation mechanism. In this work, the secondary structure of proteins in SPSS and SSS was determined by FTIR spectroscopy; the FTIR spectra including their deconvolution in infrared amide I bands (1,600–1,700 cm−1) and proportions of α-helix, β-sheet, β-turn, and random coil were shown in Fig. 4; Table 3, respectively. Significant differences, especially the key regions including amide I bands (1,600–1,700 cm−1) and amide III bands (1,200–1,300 cm−1), were observed in the FTIR spectra of proteins in SPSS and SSS. Meanwhile, the deconvolutions in infrared amide I bands (1,600–1,700 cm−1) of proteins in SPSS and SSS were used to calculate the compositions of the proteins’ secondary structure [17, 18]. The results also proved that significant differences of the secondary structure existed in proteins from SPSS and SSS. Compared with proteins in SSS, the proteins in SPSS possessed remarkably higher α-helix, β-turn and much lower β-sheet, random coil (p < 0.05). Wang et al. [27] proved that the solubility of soybean protein isolate (SPI) positively correlated with the contents of β-sheet and random coil. Liu et al. [28] and Linding et al. [29] also proved that the increase in random coil in protein was propitious to improve its solubility. In this work, the contents of β-sheet and random coil in proteins of SSS were 84.06 and 34.54 % higher than those of SPSS, respectively. The fact that proteins in SPSS were insoluble and proteins in SSS were soluble was well in accordance with the above reports. Previous report by Wang et al. [27] also showed that the solubility of SPI increased with the rise of α-helix, and no correlation was observed between the solubility of SPI and β-turn. Thus, the result that proteins in SPSS possessed significantly higher α-helix than that in SSS seemed somewhat contradictory with the above report. The much lower β-sheet and random coil in proteins of SPSS (total 29.16 % lower) when compared with SSS might be responsible for the above contradictory phenomenon. Since the solubility of protein closely correlated with its secondary structure, the significant distinction of secondary structure compositions in proteins of SPSS and SSS would lead to their difference of solubility inevitably.

The FTIR spectra of proteins from SPSS (1) and SSS (2). a is the whole spectrum band from 4,000 to 400 cm−1 in FTIR; b and c are the deconvolution in infrared amide I bands (1,600–1,700 cm−1) of proteins from SPSS and SSS. Bands 1,650.9/1,652.6 cm−1 are assigned to α-helix; bands 1,615.0/1,613.4, 1,628.0/1,627.4 cm−1 are assigned to β-sheet; bands 1,661.9/1,660.2, 1,674.0/1,671.9, 1,686.6/1,692.7 cm−1 are assigned to β-turn; bands 1,640.7/1,641.1 cm−1 are assigned to random coil [1,617, 1,718]
Conclusion
In this work, the proteins in SPSS were characterized and their formation mechanism was elucidated. Firstly, we confirmed that acidic polypeptide A1a of soy glycinin G1 (about 32–35 kDa) and basic polypeptide B3 of soy glycinin G4 (about 23 kDa) were the predominant proteins (95.2 %) in SPSS. Secondly, we proved SPSS contained not only proteins but also FAAs, and further revealed that the significantly higher average hydrophobicity and much lower contents of β-sheet and random coil were the main characteristics and formation mechanism of proteins in SPSS. Further work about the method to eliminate the secondary precipitate proteins using specific enzyme(s) is worthy to be performed.
References
Gao XL, Cui C, Ren JY, Zhao HF, Zhao QZ, Zhao MM (2011) Changes in the chemical composition of traditional Chinese-type soy sauce at different stages of manufacture and its relation to taste. Int J Food Sci Technol 46:243–249
Van der Sluis C, Tramper J, Wijffels RH (2001) Enhancing and accelerating flavour formation by salt-tolerant yeasts in Japanese soy-sauce processes. Trends Food Sci Technol 12:322–327
Kobayashi M (2005) Immunological functions of soy sauce: hypoallergenicity and antiallergic activity of soy sauce. J Biosci Bioeng 100:144–151
Kobayashi M, Magishi N, Matsushita H, Hashimoto T, Fujimoto M, Suzuki M, Tusji K, Saito M, Inoue E, Yoshikawa Y, Matsuura T (2008) Hypolipidemic effect of Shoyu polysaccharides from soy sauce in animals and humans. Int J Mol Med 22:565–570
Mashilipa C, Wang Q, Slevin M, Ahmed N (2011) Antiglycation and antioxidant properties of soy sauces. J Med Food 14:1647–1653
Gao XL, Yan S, Chen YB, Lu J (2013) Preliminary study on secondary sediment formation of soy sauces based on statistics methods. Sci Technol Food Ind 34:188–191
Zhang ZH, Li GJ, Yu SJ, Gao DW (2000) Analysis of amino acid in soy sauce and its secondary sediment. J Qiqihar Univ 16:4–7
Zhang ZH, Li GJ, Yu SJ, Gao DW (2000) Solubility of sediment in soy sauce. J South China Univ Technol (Nat Sc. Ed) 28:59–63
Fang ZX, Zhang M, Tao GJ, Sun JF, Sun JC (2006) Chemical composition of clarified bayberry (Myrica rubra Sieb. et Zucc.) juice sediment. J Agric Food Chem 54:7710–7716
Masahiro T, Asahiro T, Yoshie M, Haruo K, Yumiko Y, Kazuyoshi O (1999) Isomers and homologues of l-glutamic acid 5-n-butyl ester as promoters of sediment formation in raw soy sauce. J Biosci Bioeng 87:357–360
Tomita M, Motomura Y, Kitahara H, Yoshiki Y, Okubo K (1998) Purification and identification of the promoter of sediment formation from raw soy sauce by heating. J Biosci Bioeng 86:373–378
AOAC (1995) AOAC official methods of analysis, 16th edn. Association of Official Analytical Chemists, Arlighton
Lowry OH, Rosembroug HJ, Lewis A, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 19:265–275
Zhang XM, Shi L, Shu SK, Wang Y, Zhao K, Xu NZ, Liu SQ, Roepstorff P (2007) An improved method of sample preparation on AnchorChip targets for MALDI-MS and MS/MS and its application in the liver proteome project. Proteomics 7:340–349
Yang B, Yang HS, Li J, Li ZX, Jiang YM (2011) Amino acid composition, molecular weight distribution and antioxidant activity of protein hydrolysates of soy sauce lees. Food Chem 124:551–555
Lozano P, Combes D, Iborra JL (1994) Food protein nutrient improvement by protease at reduced water activity. J Food Sci 59:876–880
Liu YY, Zeng XA, Deng ZP, Yu SJ, Yamasaki S (2011) Effect of pulsed electric field on the secondary structure and thermal properties of soy protein isolate. Eur Food Res Technol 233:841–850
Byler DM, Brouillette JN, Susi H (1986) Quantitative studies of protein structure by FT-IR spectral deconvolution and curve fitting. Spectroscopy 1:29–32
Yuan DB, Yang XQ, Tang CH, Zheng ZX, Min W, Ahmad I, Yin SW (2009) Physicochemical and functional properties of acidic and basic polypeptides of soy glycinin. Food Res Int 42:700–706
Shen L, Tang CH (2012) Microfluidization as a potential technique to modify surface properties of soy protein isolate. Food Res Int 48:108–118
Yang HX, Wang F, Shan L, Bi YP (2003) The progress of the studies on soybean seeds protein genes and the regulation of their expression. Soybean Sci 22:296–300
Mo XQ, Zhong ZK, Wang DH, Sun XZ (2006) Soybean glycinin subunits: characterization of physicochemical and adhesion properties. J Agric Food Chem 54:7589–7593
Momma T, Negoro T, Hirano H, Matsumoto A, Udaka K, Fukazawa C (1985) Glycinin A5A4B3 mRNA: cDNA cloning and nucleotide sequencing of a splitting storage protein subunit of soybean. Eur J Biochem 149:491–496
Zarkadas CG, Gagnon C, Gleddie S, Khanizadeh S, Cober ER, Guillemette RJD (2007) Assessment of the protein quality of fourteen soybean [Glycine max (L.) Merr.] cultivars using amino acid analysis and two-dimensional electrophoresis. Food Res Int 40:129–146
Bigclow CC (1967) On the average hydrophobicity of proteins and the relation between it and protein structure. J Theor Biol 16:187–211
Niwa T, Ying BW, Saito K, Jin WZ, Takada S, Ueda T, Taguchi H (2009) Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc Natl Acad Sci USA 106:4201–4206
Wang C, Jiang LZ, Wei DX, Li Y, Sui XN, Wang ZJ, Li D (2011) Effect of secondary structure determined by FTIR spectra on surface hydrophobicity of soybean protein isolate. Proc Eng 15:4819–4827
Liu RL, Li XH, Zhou XL, Yu J, Wang FX, Wang JH (2011) Effects of glutaminase deamidation on the structure and solubility of rice glutelin. LWT-Food Sci Technol 44:2205–2210
Linding R, Schymkowitz J, Rousseau F, Diella F, Serrano L (2004) A comparative study of the relationship between protein structure and β-aggregation in globular and intrinsically disordered proteins. J Mol Biol 342:345–353
Acknowledgments
The authors gratefully acknowledge the following programs for their financial supports: the Major State Basic Research Development Program of China (973 Program, 2012CB720802), the Open Project of Key Laboratory of Industrial Biotechnology, Ministry of Education, Jiangnan University (KLIB-KF201108), the Fundamental Research Funds for the Central Universities, Jiangnan University (JUSRP111A26) and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Conflict of interest
None.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Gao, X., Sun, P., Lu, J. et al. Characterization and formation mechanism of proteins in the secondary precipitate of soy sauce. Eur Food Res Technol 237, 647–654 (2013). https://doi.org/10.1007/s00217-013-2054-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-013-2054-2