
Abstract
In this study, 3′-flanking sequence between the host plant DNA and the integrated gene construct of pHMW1Dx5 vector in transgenic wheat B73-6-1 was revealed by means of adaptor PCR; thus, the fragment with the length of 3.1 kb was obtained, including a 190-bp wheat genomic DNA, which demonstrates that this HMW-GS gene was located on the wheat chromosome 3B. And the event-specific PCR primers were designed based upon the revealed 3′-flanking sequence; the conventional qualitative PCR and quantitative SYBR real-time PCR detection methods employing these primers were successfully developed. In conventional qualitative PCR assay, the limit of detection was 0.1 % for B73-6-1 wheat genomic DNA for one reaction. In the quantitative SYBR real-time PCR assay, the limit of detection and limit of quantification were 10 and 100 haploid genome copies, respectively. In addition, three mixed blind wheat samples with known B73-6-1 contents were detected using the established real-time PCR systems, and the ideal results indicated that the established event-specific real-time PCR detection systems were reliable, sensitive and accurate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nowadays, genetically modified organisms (GMOs) have been widely developed and applied in agriculture, a 94-fold increase in hectarage from 1.7 million hectares in 1996 to 160 million hectares in 2011 makes biotech crops the fastest adopted crop technology in the history of modern agriculture [1]. To strengthen the regulation of GMOs, more than 50 countries and areas have published a series of laws and rules for GMO regulation and labeling [2]. Since labeling of foods containing GMO ingredients is mandatory in some countries, the demand to develop easy and reliable detection methods for GMOs is very high [3].
Wheat is the most important crop in the world in terms of the area under cultivation, yield and geographic distribution. It is widely used in food processing (e.g., for bread, pasta, noodles) and as feed for livestock [4]. Wheat flour is different from other cereal flours, including maize, because it contains gluten that gives it the elasticity and extensity required for bread making [5]. Gluten consists mainly of two types of seed-storage proteins, the glutenin and the gliadin. Glutenin are classified into high-molecular-weight (HMW) subunits and low-molecular-weight (LMW) subunits. The elasticity of wheat dough depends mainly on the HMW glutenin, so they are important determinants of bread-making quality [6].
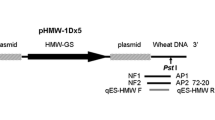
The transgenic wheat B73-6-1 expressing an increased amount of a HMW glutenin subunit (HMW-GS gene) was obtained by co-transformation of the plasmid vectors pHMW1Dx5 (carrying HMW-GS gene which was driven by its own specific promoter) and pAHC25 (carrying selectable and screenable marker genes Bar and GUS driven by the ubiquitin promoter), which results in B73-6-1 is different from other GM crops. The transgenic line B73-6-1 has been confirmed by large-scale testing of grain grown in field trials [7] and has passed the intermediate testing phase followed by the environmental release phase. So, it is representative of the type of transgenic wheat which may grow commercially in the future. In this study, based on the 3′-flanking sequence of plasmid pHMW1Dx5 containing a 8.4-kb genomic fragment composed of the coding sequence of the Glu-D1-1b Gene, 3.8 kb 5′ UTR and 2.2 kb 3′ UTR [8], the 3′ integration flanking sequence of pHMW1Dx5 vector in B73-6-1 was revealed by genome walking, and the event-specific qualitative and quantitative detection methods were established.
Materials and methods
Plant materials
GM wheat B73-6-1 and B72-8-11 were kindly supplied by Heilongjiang Entry-Exit Inspection and Quarantine Bureau. GM soybean (MON89788 and GTS 40-3-2), GM maize (MON810 and MON863) and GM cotton (MON531 and MON1445) were developed and supplied by Monsanto Company. Bt176 was developed and supplied by Syngenta Seeds, Inc., GM rice (TT51-1 rice and “Kefeng 6”) were kindly provided by China National Rice Research Institute. GM canola (MS1, RF3, MS8), Liberty Link soybean A2704-12 and GM cotton LLCotton25 were developed and supplied by Bayer BioScience. Non-transgenic wheat and rice seeds were purchased from local market in Harbin.
Oligonucleotide primers
Sequences of oligonucleotide primers employed in this study are listed in Table 1 and were designed using Primer Premier 5 (PREMIER Biosoft Int., Palo Alto, CA).The specific primers HF6761, HF8323, HFd495, HFd832, HFd2280 and HFd2438 (Table 1) used in adaptor PCR were designed based on the sequence of the pHMW1Dx5 plasmid, and the adaptor primers AP1 and AP2 were also selected and used in adaptor PCR. The event-specific PCR primer pairs (HMW F/R and qHMW F/R) used for B73-6-1 wheat detection were designed based on the flanking region between the host DNA and the exogenous sequence originating from the pHMW1Dx5 vector, one of the two co-transformation vectors, and the primers HMW F/R were employed for conventional PCR, that is, qualitative PCR, and the primers qHMW F/R were employed for SYBR Green I real-time PCR (Table 1). For the development of the event-specific detection method of B73-6-1 wheat, various endogenous reference genes were used in conventional and real-time PCR [9–14], respectively. All of the primers were synthesized and purified by Invitrogen (Beijing, China).
DNA extraction and construction of genome walking libraries
Total genomic DNA was isolated from seeds by the CTAB method. The quantity of DNA samples was calculated using absorbance measurements at 260 nm wavelength. Four experimental libraries (Pvu I, Dra I, Stu I and EcoRV) were constructed using Genome Walker™ Universal Kit according to the procedures described in the manufacturer instructions.
Genome walking and verification of the wheat DNA fragment in flanking sequence
The 3′-flanking sequence of pHMW1Dx5 vector in B73-6-1 wheat was identified following the instruction manual (CLONTECH). The sequences of all primers are shown in Table 1.
The amplified fragments in the adaptor PCR amplifications were purified and cloned into pMD18-T vector (TaKaRa Biotechnology Co., Ltd.). Fully sequencing analysis of the PCR products was performed by BGI (Beijing Genomics Institute, China).
In order to validate the wheat genomic DNA fragment derived from wheat B73-6-1 3′-flanking sequence, the primers of WG (wheat genome) F/R (Table 1) were designed according to the 3′-flanking sequence obtained from the libraries. In PCR assay, the amplifications were carried out in 20 μL volume reactions, with 50 ng of genomic DNA, 1 × PCR buffer, 0.25 mM dNTP mix, 0.2 μM WG F/R primer and 1 unit of rTaq DNA polymerase (TaKaRa Biotechnology Co., Ltd.). The PCR amplifications were performed in Tgradient (Biometra, Göttingen, Germany) with the program as follows: one step of 5 min at 95 °C, 35 cycles of 30 s at 95 °C, 30 s at 58 °C, 30 s at 72 °C; one step of 5 min at 72 °C. Amplification products were electrophoresed in 2 % agarose gels for approximately 20 min at 100 V. Figure 1 shows the primer location within the B73-6-1 3′-flanking sequence.

Digestion of wheat genome using four restriction enzymes of Pvu I, Dra I, Stu I and EcoR V. 1 DL2000 DNA marker, 2–5 the B73-6-1 wheat genome were completely digested by the restriction enzymes of Pvu I, Dra I, Stu I and EcoR V, respectively
Qualitative PCR assay
In qualitative PCR assays, all of the amplifications were carried out in 20 μL volume reactions, with 100 ng of genomic DNA, 1 × PCR buffer, 0.25 mM dNTP mix, 0.2 μM GAG56D F/GAG56D R (house-keeping gene) HMW F/R (event-specific gene) (Table 1) and 1U rTaq DNA polymerase. All the qualitative PCR and electrophoresis were conducted as described above in Tgradient. Each reaction of one test was repeated three times and each time with triple replications.
The estimation of the specificity was carried out on different crops (including B73-6-1), 16 kinds of GM crops and 2 kinds of non-GM crops using the HMW F/R primers for exogenous gene and different house-keeping genes (Table 1).
For LOD tests, the seeds of B73-6-1 wheat and the non-transgenic wheat were mixed to produce the mixed sample containing relative amounts of B73-6-1 of 10, 5, 2, 1, 0.5, 0.1, 0.05 and 0.01 % (m/m). The mixed samples were used to extract DNA, and each sample was independently divided in quadruplicate for DNA extraction. DNA extracts containing different percentage of transgenic materials were used for assessing the limit of detection with the pair of primer HMW F/R.
The reproducibility of the developed method was validated by three researchers.
Quantitative PCR assay
For event-specific PCR assay, primers qHMW F/R leading to a 163-bp product were used. PCR contained 1 × SYBR® Premix Ex Taq™ II (Takara, Otsu, Japan), 200nM primers, 1 × ROX reference dye and 1 μl of the DNA solution. Real-time PCR were performed using the following program: 30 s 95 °C, 40 cycles of 5 s at 95 °C and 31 s at 60 °C. After PCR amplification, Tm curve analysis was performed described as followed: the PCR products were heated to 95 °C during 15 s, cooled at 60 °C for 20 s and then slowly heated to 95 °C at a rate of 0.2 °C/s. Standard curves for event specific and wx012 assays were established with five dilutions of DNA from B73-6-1 wheat. Genomic DNA of 100 % B73-6-1 wheat was serially tenfold diluted with ddH2O to final concentrations equivalent to 100,000, 10,000, 1,000, 100, 10 and 1 copies of haploid genome/μL, 1 μL of diluted DNA sample was added, and all reactions were repeated three times, each time with triple parallels for each template DNA.
Result
Characterization of the 3′-flanking sequence of pHMW1Dx5 vector in B73-6-1 wheat
Quality of the purity of genomic DNA by restriction endonuclease digestion
To check the digestion products by restriction endonucleases of Stu I, Dra I, Pvu I and EcoR V, 5 μl of each reaction mixture was loaded on 1 % agarose gel in 1 × TAE, and the smear indicated that the DNA could be digested completely by restriction enzymes (Fig. 1) and be used for genome walking.
Characterization and cloning of 3′-flanking sequence of the B73-6-1 wheat
In order to obtain the 3′-flanking sequence pHMW1Dx5 vector in B73-6-1, a modified adaptor PCR method based on the 3′ junction characterization method described by Genome Walking Kit user manual was used. Adaptor PCR amplifications were performed with combinations of different primers of the HMW-GS gene using B73-6-1 and non-GM wheat genomic DNA as templates. In the three nested PCR, 3 discrete flanking fragments were amplified by the combination of the primers AP1/GSP1 (the gene-specific primers for primary PCR) and AP2/GSP2 (the gene-specific primer for secondary PCR) (Table 1, Fig. 2), and the amplification results are shown in Fig. 3.

Schematic diagram of GM wheat B73-6-1-integrated DNA. “73–29” of 1.2 kb length was amplified in the first nested PCR using Dra I-digested DNA as template with the primers AP1/HF6761 and AP2/HF8323. In the second nested PCR, “73–35” of approximately 2.0 kb length was amplified with Pvu I-digested DNA using the primers AP1/HFd495 and AP2/HFd832. And the third discrete product “73–37” with the length of about 500 bp was amplified using the genomic DNA digested by EcoR V as the template. The primer pairs of HMW F/R, qHMW F/R and WG F/R were used to amplify the GM wheat B73-6-1 event-specific fragment and wheat genomic conservative fragment, respectively

PCR amplification results of identification of HMW-GS 3′-flanking sequence with B73-6-1 DNA. M, DL2000 marker; 1 product of 73–29 with primers of AP2/HF8323, 2 product of 73–35 with primers of AP2/HFd832, 3 product of 73–37 with primers of AP2 and HFd2438
The 3′ site of B73-6-1 event integration locus on wheat genome sequence
The 3′-flanking sequence in wheat B73-6-1 was determined using the adaptor PCR method, which is comprised of nested PCR. In the first nested PCR, one discrete PCR product named as “73–29” of 1.2 kb length was amplified using Dra I-digested DNA as template with the primers AP1/HF6761 and AP2/HF8323 (Fig. 3), which originated from backbone sequence of cloning vector pVLG6 and has no homologous to wheat chromosome through blast search in NCBI database. The 1,286-bp DNA sequence consisted of two parts, one was a 134-bp sequence of NOS terminator region and the other was an 1,152-bp sequence of cloning vector (Fig. 4a). Since “73–29” could not locate the HMW-GS insertion site on the wheat chromosome, the second nested PCR was performed using the primers AP1/HFd495 and AP2/HFd832; consequently, PCR product named as “73-35” of approximately 1.9 kb length was amplified with Pvu I-digested DNA (Fig. 3). The sequencing result showed that one 1,878-bp DNA fragment encompassing the 3′ junction region was obtained, which consisted of one 742-bp sequence of cloning vector and one 1,136-bp sequence (Fig. 4a) of which was identical to the sequences of E. coli str. K12 substr. DH10B genome and showed 100 % homology with its sequence. But the blast search of this fragment still showed no homologous sequence existed on the wheat genomic DNA. Furthermore, the third nested PCR primers (AP1 and HFd2280, AP2 and HFd2438) were designed according to the sequence of “73–35” to amplify the third discrete sequences. And the third discrete product “73–37” was amplified with the length of about 500 bp using the genomic DNA digested by EcoR V as template (Fig. 3). The result of sequence analysis indicated that one 536-bp fragment was obtained, of which 263 bp originated from E. coli str. K12 substr. DH10B genome, 190 bp originated from wheat genomic DNA and 83-bp DNA fragment with low homology to known sequences (Fig. 4a). This 190-bp DNA fragment showed 82 % homology with the sequence in the untranslated region (UTR) of BAC library from wheat chromosome 3B (Genbank accession number FN5644289) (Fig. 4b).

The sequence of B73-6-1 3′-flanking sequence and the locations of all primers. Capital letters represent the sequence from the cloning vector sequence and E. coli str. K12 substr. DH10B genome, and lowercase letters show the flanking wheat genomic sequence (FN564428). The position of the GSP1, GSP2, event specific and wheat genomic DNA primers are highlighted in bold and indicated by arrows. The hollow arrow shows the junction sites between the insert and the host DNA. The ellipsis represents the omitted sequence of the 3′-flanking sequence. Dotted arrows indicate the positions of primers WG F/R, HMW F/R and qHMW F/R, respectively
Identification of the wheat genomic DNA in the flanking fragment
To further identify the wheat chromosome 3B DNA of the flanking sequence, we continued to design the primers WG F/R (Table 1) to amplify the product directly from B73-6-1, B72-8-11 and non-GM wheat flour. The 169-bp fragment was amplified from all the wheat materials (Fig. 5), and the location of the primer WG F/R is shown in Figs. 2 and 4a. These results demonstrated that the flanking sequence indeed contained the sequence of wheat chromosome 3B genome DNA.

Verification of the flanking fragment using WG F/WG R primers. 1 DL2000 marker, 2 NTC, 3 non-GM wheat, 4 B73-6-1, 5 B72-8-11
Qualitative detection of B73-6-1 wheat by conventional PCR
Development of the event-specific PCR detection method for B73-6-1 wheat
The primers HMW F/R were designed based on the 3′-flanking sequence described above and employed to establish the qualitative event-specific PCR assay for B73-6-1 wheat (Fig. 5). A specific amplification product of 388 bp was obtained as expected when using B73-6-1 genomic DNA as a template, but no amplification was observed with the non-GM wheat DNA and B72-8-11 template (Fig. 6).

Specificity detection of the event-specific qualitative PCR method. 1 DL2000 DNA marker, 2 non-GM wheat, 3 B73-6-1, 4 B72-8-1, 5 Bt176, 6 MON810, 7 MON863, 8 Roundup Ready soy, 9 MON89788, 10 A2704-12, 11 TT51-1, 12 Kefeng 6, 13 non-GM rice, 14 MON531, 15 MON1445, 16 LLCOTTON25, 17 MS1, 18 MS8, 19 RF1
Specificity of the qualification assays
When used in an event-specific PCR with B73-6-1 DNA as a template, a single fragment of approximately 388 bp was observed as expected (Fig. 6). No amplification was detected using B72-8-11, Bt176, MON810, MON863, GTS40-3-2, MON89788, A2704-12, TT51-1, Kefeng 6, non-GM rice, MON531, MON1445, LLCOTTON25, MS1, MS8, RF1 and non-GMO wheat DNA as templates in the assay, indicating the specificity of the PCR. In this study, six primer pairs of different house-keeping genes of the crops were performed and were successfully used to amplify all the selected GM crops (Fig. 7). The product length of the house-keeping genes were as follows: 319 bp GAG56D gene for wheat, 88 bp zSSIIb gene for maize, 118 bp lectin gene for soybean, 277 bp SPS gene for rice, 107 bp Sad1 gene for cotton, and 206 bp HMG I/Y gene for canola.

PCR amplification among the 6 kinds of house-keeping genes. 1 DL2000 marker, 2–4 the primers of GAG56D for non-GM wheat, B73-6-1, B72-8-1, 5–7 the primers of zSSIIb for Bt176, MON810, MON863 maize, 8–10 the primers of lectin for Roundup Ready soy, MON89788, A2704-12 soy, 11–13 the primers of SPS for TT51-1, Kefeng 6, non-GM rice, 14–16 the primers of Sad1 for MON531, MON1445, LLCOTTON25 cotton, 17–19 the primers of HMG I/Y for MS1, MS8, RF1
Assessment of the LOD
In qualitative PCR, the testing sensitivity was measured by the LOD method. The results showed that the target fragment was detected at all of the testing levels tested except the 0.05 % and 0.01 % levels, which meant that the lowest testing level was 0.1 % (Fig. 8).

Sensitivity of B73-6-1 event-specific qualitative detection. 1 DL2000 DNA marker, 2–9 10, 5, 2, 1, 0.5, 0.1, 0.05, 0.01 % B73-6-1 contents, respectively, 10 NTC
Reproducibility of the qualification PCR assays
Repeatability and reproducibility of the qualification PCR was determined using the B73-6-1 DNA three parallels and three replications (Fig. 9), and the results of the repeatability and reproducibility tests indicated that the B73-6-1 event-specific qualitative PCR assay was reliable in B73-6-1 wheat qualification.

Reproducibility of B73-6-1 event-specific qualitative detection. 1 DL2000 DNA marker, 2–4 the 3 samples of the first amplification, 5–7 the 3 samples of the second amplification, 8–10 the 3 samples of the third amplification
Quantitative detection of B73-6-1 by SYBR real-rime PCR
Construction of standard curves
To evaluate the B73-6-1 event-specific real-time PCR assay and GM B73-6-1 wheat sample quantification, a series of B73-6-1 wheat genomic DNA dilutions were used for the test of reproducibility and repeatability, determination of the limits of detection and quantification (LOD and LOQ) and construction of standard curves. The real-time PCR amplified results showed that the PCR efficiency was = 0.96, and the squared regression coefficient (R 2) of the standard curve was 0.996 (Fig. 10). The high PCR efficiency indicated that this real-time PCR assay was suitable for B73-6-1 genomic DNA quantification. The good linearity between DNA quantities and fluorescence values (Ct) indicated that these assays were well suitable for quantitative measurements. y = −2.246x + 38.233.

Amplification plots and standard curve for B73-6-1 event-specific real-time PCR assay. a Amplification curves (serial DNA dilutions corresponding to 100,000, 10,000, 1,000, 100 and 10 copies of B73-6-1 haploid genome per reaction) were generated for B73-6-1 wheat quantification; (b) parameters of the regression line through data points are indicated within the standard curve
Repeatability and reproducibility
The reproducibility and repeatability of real-time PCR assays can be analyzed by the data shown in Table 2. For the real-time PCR assay for event-specific gene detection, the mean Ct values varied from 24.76 to 33.61 with a standard deviation (SD) value from 0.20 to 0.48 and coefficient of variation (CV) value from 0.79 to 1.40 %. These results indicating that quantitative PCR assays established were stable and reliable.
Limits of detection and quantification (LOD and LOQ)
As expected, the ability to detect B73-6-1 wheat decreased with decreasing genomic DNA copy numbers, and 10 copies of B73-6-1 genomic DNA have been detected seven times in a total of nine repeated reactions. However, when the template copy number was 1, no amplicons could be detected in 9 replicates. The results indicated that the LOD value was about 10 copies. The data also showed that the SD values of the nine reactions with the same template concentration increased with decreasing copy number. To obtain reliable quantization results under ideal conditions, approximately 100 initial template copies were required, and we concluded that the LOQ of the event-specific real-time PCR assay was 100 copies of the haploid genome.
Blind sample quantification
Mixed samples were prepared to evaluate the accuracy and precision of the established real-time PCR methods in this study. GM B73-6-1 wheat DNA samples S1, S2 and S3 with 6, 3 and 1 %, respectively, were artificially prepared by mixing the pure B73-6-1 DNA with non-GM wheat genomic DNA on a genome/genome basis and were used for quantification in developed B73-6-1 event-specific PCR assay. The mean values of three replicates for each sample as provided by all participants are shown in Table 3. The mean quantitative results of these three DNA samples (S1, S2, and S3) were 5.95, 3.10 and 1.03 %, respectively. The average bias between quantified values and true values of blind samples were 0.88, 3.20 and 3.39 %, respectively. These results showed that the bias values of practical samples were lower than the acceptance threshold of 25 % of one GMO detection method [16], indicating that the developed B73-6-1 real-time PCR assay is creditable and suitable for the quantification of GM B73-6-1 wheat and its derivates.
Discussion
Various PCR-based methods are available for chromosome walking from a known sequence to an unknown region [17]. Several PCR-based methodologies are available for walking from a known region to cloned or uncloned genomic DNA, including inverse PCR (IPCR), adaptor ligation-mediated PCR (LM-PCR) and randomly primed PCR [18]. In addition, the flanking sequences of more and more GM crops are also being identified for the development of event-specific detection method. In this study, adaptor ligation-mediated PCR was performed to identify the flanking sequence of B73-6-1, and three junction regions of 73-29, 73-35 and 73-37 were isolated. After the splicing of 73-29, 73-35 and 73-37, a discrete product of approximately 3.1 kb was obtained (Fig. 4). The search result using blast in the NCBI database showed that the 3.1-kb assembled sequence of the 3′-flanking sequence contained 134 bp originating from the NOS terminator, 1.6 kb originating from the universal sequence of cloning vector, 1.1 kb originating from E. coli DNA and 190 bp originating from endogenous wheat genomic DNA. This 190-bp sequence was identified as a part of the loci from 1291474 to 1291675 in the wheat 3B-specific chromosome (Genbank Accession number FN564428). No amplification product was observed in parallel experiments using B72-8-11 and non-GM wheat as templates, which showed that the event specificity of this flanking sequence. But we have no idea how to explain the existence of E. coli genome, and we speculate that it is the result of homologous recombination. To further demonstrate that this 190-bp fragment did indeed correspond to wheat flanking sequences, we used this wheat genomic sequence to design primer WG F/R and performed standard PCR with this primer pair using non-GM wheat, B73-6-1 and B72-8-11 DNA as templates. As shown in Fig. 5, an amplification product of approximate size 169 bp was obtained with non-GM wheat DNA, B73-6-1 and B72-8-11. The PCR product was cloned into the pMD18T vector and sequenced. These data suggested that a unique transgene element of about 3.1 kb was present in the genome of wheat B73-6-1.
PCR-based GMO tests can be grouped into at least four categories, such as screening, gene-, construct- and event-specific PCR [2]. The four PCR detection systems were developed based on the four different kinds of target DNA fragments of exogenous integration corresponding to their level of specificity. Each category corresponds to the composition of the DNA fragment that is amplified in the PCR. The flanking sequence is the only unique signature of a transformation event (within the limitations of present day technology), which is the junction at the integration locus between the recipient genome and the inserted DNA. This junction is the target of category of event-specific method. Currently, the event-specific PCR method has been the key trends for GMO identification and quantification because of its high specificity based on the flanking sequence [19, 20]. Nowadays, only screening PCR of qualitative PCR [9] and quantitative PCR [21] was developed for amplification of the endogenesis genes of GAG56D and wx012 and extrinsic genes of ubiquitin promotor, NOS terminator, Bar and GUS reporter gene, and no event-specific PCR method was used to detect B73-6-1 as far as we know. Currently, the qualitative or quantitative event-specific detection methods have been used in the detection of GM crops, such as the GTS 40-3-2 soybean [22–24], MON810 [25], MON863 [26, 27], MON89788 [2], MON88913 [28], NK603 [29], GT73 [30], GA21 [31], T25 [32], Carnation Moonlite [33], Maize 59122 [34], Kefeng 6 [35] and so on.
The event-specific qualitative PCR system was developed to detect the wheat B73-6-1, and the primer HMW F/R was located at the B73-6-1 3′-flanking sequence (Figs.2 and 4a). The wheat GAG56D gene was selected as an endogenous control gene, and the primer pair GAG56D F/R was employed in PCR assay of wheat identification. As expected, in the established qualitative PCR assay, one single 388-bp DNA fragment was obtained using the B73-6-1 wheat DNA as template, and no fragment was detected in other GMOs and no template control (NTC) (Fig. 6). The target fragments of endogenous genes were individually detected in all GM wheat, maize, soy, rice, cotton, canola and non-GM crops except for NTC (Fig. 7). These data confirmed that the obtained DNA fragment was a B73-6-1 event-specific region between the exogenous DNA and the wheat genomic DNA and indicated that the sequence was unique to B73-6-1 and thus could be suitable for use in event-specific PCR assay. The two controls of non-GM wheat and B72-8-11 are important, as rearrangements are known to occur during insertion of transgenic DNA in plants. The HMW F/R primer pair may form the basis for a qualitative test for presence or absence of B73-6-1 wheat DNA. So the method developed in this article is directly detect the HMW gene (target gene) in the pHMW1Dx5 vector with more feasibility and rationality than the event-specific qualitative detection method, which was constructed for the Bar gene (marker gene) in the pAHC25 based on the 3′-flanking sequence of Bar gene [36]. Additionally, the quantitative PCR detection technique was established for enriching the event-specific method of B73-6-1.
For one ideal qualitative PCR assay, a high LOD is very important and necessary, especially for the PCR assays of GMO detection. To test the LOD of the established event-specific PCR assay, DNAs extracted from different B73-6-1 wheat mixed with non-GM wheat flours were used as templates. The results indicate that the established qualitative event-specific PCR detection assay of GM wheat B73-6-1 is suitable for the practical detection of GM wheat samples. These LODs were similar to those of GM soybean [23] and GM maize [26], which indicates that the established qualitative event-specific PCR detection system of GM wheat B73-6-1 is suitable for the practical detection of GM wheat samples with high specificity and little labor.
The SYBR primers (qHMW F/R) based on the revealed 3′-flanking sequence were designed and employed to develop the event-specific real-time PCR assay for B73-6-1 wheat (Figs. 2 and 4a). Considering the haploid genome size of wheat was estimated to be 16,000 Mbp, corresponding to a molecular weight of 16.6 pg (assuming that 965 Mb weigh 1 pg) [25], the range of copy numbers from 100,000 to 1 was sufficient to quantify GMOs from 0.01 to 100 % in the 200 ng of the template for one reaction. The range was supposed to be sufficient to quantify GMOs because of the quantified threshold values of labeling regulations in EU (0.9 %), Korea (3 %) and Japan (5.0 %). For development of the B73-6-1 event-specific real-time PCR assay, the quantitative standard curve, repeatability and reproducibility, and LOD or LOQ were performed using six serial diluted concentrations of B73-6-1 wheat genomic DNA were used for the preparation of standard curves, containing approximately 100,000–10 copies of haploid genome per reaction, while when the template copy number was 1, no amplicons could be detected in the template with one copy haploid genomes in nine repeated reactions. To obtain reliable quantification results under ideal conditions, approximately 100 initial template copies were required, and we concluded that the LOQ of the event-specific real-time PCR assay was 100 copies of haploid genome.
In conclusion, we reported the 3′-flanking sequence of GM wheat B73-6-1 and located the HMW-GS gene on the wheat genome. Based on this sequence, we established the event-specific qualitative and quantitative PCR systems for the reliable and accurate detection of B73-6-1 wheat. Due to the specificity of the established systems, we believe that this method is a new contribution to the labeling system for GMOs and also suitable for qualitative and quantitative detection of food and feed products in the future.
References
James C. (2011) Global status of commercialized biotech/GM crops: 2011. ISAAA brief no. 43. ISAAA, Ithaca
Liu J, Guo JC, Zhang HB, Li N, Yang LT, Zhang DB (2009) Development and in-house validation of the event specific polymerase chain reaction detection methods for genetically modified soybean MON89788 based on the cloned integration flanking sequence. J Agric Food Chem 57:10524–10530
Windels P, Taverniers I, Depicker A, Bockstaele EV, Loose MD (2001) Characterisation of the Roundup Ready soybean insert. Eur Food Res Technol 213:107–112
Alvarez ML, Guelman S, Halford NG, Lustig S, Reggiardo MI, Ryabushkina N, Shewry P, Stein J, Vallejos RH (2000) Silencing of HMW glutenins in transgenic wheat expressing extra HMW subunits. Theor Appl Genet 100:319–327
Barro F, Rooke L, Bekes F, Gras P, Tatham AS, Fido R (1997) Transformation of wheat with high-molecular-weight subunit genes results in improved functional properties. Nature Biotechnol 15:1295–1299
Shewry PR, Halford NG, Tatham AS, Popineau Y, Lafiandra D, Belton PS (2003) The high molecular weight subunits of wheat glutenin and their role in determining wheat processing properties. Adv Food Nutr Res 45:221–302
Darlington H, Fido R, Tatham AS, Jones HD, Salmon SE, Shewry PR (2003) Milling and baking properties of field grown wheat expressing HMW subunit transgenes. J Cereal Sci 38:301–306
Anderson OD, Greene FC, Yip RE, Halford NG, Shewry PR, Malpica-Romero JM (1989) Nucleotide sequences of the two high-molecular-weight glutenin genes from the d-genome of a hexaploid bread wheat Triticum aestivum L. cv Cheyenne. Nucl Acids Res 17:461–462
Luan FX, Zhang HX, Bai Y (2007) Screening and detecting of genetically modified components in transgenic wheat by PCR. J Triticeae Crops 27:378–381 (In Chinese)
Bulletin No.869-8-2007 of the Ministry of Agriculture of the People’s Republic of China (2007) Detection of genetically modified plants and derived products qualitative PCR method for insect-resistant maize MON810 and its derivates[S]. China Agriculture Press, Beijing
Bulletin No.1485-6-2010 of the Ministry of Agriculture of the People’s Republic of China (2010) Detection of genetically modified plants and derived products qualitative PCR method for herbicide-tolerant soybean MON89788 and its derivates[S]. China Agriculture Press, Beijing
Bulletin No.1193-3-2009 of the Ministry of Agriculture of the People’s Republic of China (2009) Detection of genetically modified plants and derived products qualitative PCR method for insect-resistant rice TT51-1 and its derivates[S]. China Agriculture Press, Beijing
Bulletin No.1485-10-2010 of the Ministry of Agriculture of the People’s Republic of China (2010) Detection of genetically modified plants and derived products qualitative PCR method for herbicide-tolerant cotton LLcotton25 and its derivates[S]. China Agriculture Press, Beijing
Bulletin No.869-4-2007 of the Ministry of Agriculture of the People’s Republic of China (2007) Detection of genetically modified plants and derived products qualitative PCR method for herbicide-tolerant rapeseed MS1, RF1 and their derivates[S]. China Agriculture Press, Beijing
Luan FX, Zhang HX, Bai Y (2007) Protocol of polymerase chain reaction and real-time PCR for qualitative detecting genetically modified components in transgenic wheat[S]. SN/T 1943–2007 Standards Press of China, Beijing
Definition of Minimum Performance Requirements for Analytical Methods of GMO Testing-ENGL, http://gmo-crl.jrc.ec.europa.eu/doc/Min_Perf_Requirements_Analytical_methods.pdf
Liu B, Su Q, Tang MQ, Yuan XD, An LJ (2006) Progress of the PCR amplification techniques for chromosome walking. Hereditas 28:587–595
Zhang Y, Liu DQ, Yang WX, Li YN, Yan HF (2011) A nonspecific primer anchored PCR technique for chromosome walking. Brazilian Arch Biol Andtechnol 54:99–106
Holst-Jensen A, Røning SB, Løseth A, Berdal KG (2003) PCR technology for screening and quantification of genetically modified organisms (GMOs). Anal Bioanal Chem 375:985–993
Miraglia M, Berdal KG, Brera C, Corbisier P, Holst-Jensen A, Kok EJ et al (2004) Detection and traceability of genetically modified organisms in the food production chain Food Chem. Toxicology 42:1157–1180
Bai Y, Luan FX, Wang X (2009) Detection Limits of PCR and Real time PCR for Genetically Modified Components in Transgenic Wheat. J Triticeae Crop 29:199–205 (In Chinese)
Taverniers I, Windels P, Van BE, De M (2001) Use of cloned DNA fragments for event specific quantification of genetically modified organisms in pure and mixed food products. Eur Food Res Technol 213:417–424
Lau LT, Collins RA, Yiu S, Xing J, Yu AC (2004) Detection and characterization of recombinant DNA in the Roundup Ready soybean insert. Food Control 15:471–478
Terry CF, Harris N (2001) Event specific detection of roundup ready soya using two different real time PCR detection chemistries. Eur Food Res Technol 213:425–431
Hernández M, Pla M, Esteve T, Prat S, Puigdomènech P, Ferrando A (2003) A specific real-time quantitative PCR detection system for event MON810 in maize YieldGard® based on the 3′-transgene integration sequence. Transgenic Res 12:179–189
Yang LT, Pan AH, Zhang KW et al (2005) Qualitative and quantitative PCR methods for event specific detection of genetically modified cotton Mon1445 and Mon531. Transgenic Res 14:817–831
Pan AH, Yang LT, Xu SC, Yin CS, Zhang KW, Wang ZY et al (2006) Event specific qualitative and quantitative PCR detection of MON863 maize based upon the 3′-transgene integration sequence. J Cereal Sci 43:250–257
Yang LT, Jiang LX, Shen KL, Guo JC, Rao J, Seonghun L, Zhang DB (2009) Event specific qualitative and quantitative PCR detection methods for genetically modified cotton MON88913. Food Safety Qual Detect Technol 1:10–19
Nielsen CR, Berdal KG, Holst JA (2004) Characterisation of the 5′ integration site and development of an event specific real-time PCR assay for NK603 maize from a low starting copy number. Eur Food Res Technol 219:421–427
Yang R, Xu WT, Luo YB, Guo F, Lu Y, Huang KL (2007) Event specific qualitative and quantitative polymerase chain reaction detection of Roundup Ready event GT73 based on the 3′-transgene integration sequence. Plant Cell Repots 26:1821–1831
Hernández M, Esteve T, Prat S, Pla M (2004) Development of real-time PCR system based on SYBR Green I, Amplifluor and TaqMan technologies for specific quantitative detection of the transgenic maize event GA21. J Cereal Sci 39:99–107
Cécile C, Alexandra S, Georges B, Francine B, Géraldine C, Annick D et al (2005) Characterization and event specific-detection by quantitative real-time PCR of T25 maize insert. J AOAC Int 88:536–546
Li P, Pan AH, Jia Jun W, Jiang LX, Bai L, Tang XM (2010) Establishment of event specific qualitative PCR for detecting transgenic carnation moonlite. J Agric Sci Technol 12:109–113
Xu WT, Yang R, Lu J, Zhang N, Luo YB, He J et al (2011) Event specific transgenic detection of genetically modified maize 59122 with flanking sequence. Food Sci 32:139–142 (In Chinese)
Wang WX, Zhu TH, Lai FX, Fu Q (2011) Event-specific qualitative and quantitative detection of transgenic rice Kefeng-6 by characterization of the transgene flanking sequence. Eur Food Res Technol 232:297–305
Nan Huo, Minghui Zhang, Ying Liu et al (2011) Establishment of even specific qualitative PCR for detecting genetically modified wheat B73–6-1. China Biotechnol 10:100–105 (In Chinese)
Acknowledgments
This study was supported by the foundation of Educational Committee of Heilongjiang Province of China (No.12521018). This is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, M., Huo, N., Liu, Y. et al. Event-specific detection of genetically modified wheat B73-6-1 based on the 3′-flanking sequence. Eur Food Res Technol 235, 1149–1159 (2012). https://doi.org/10.1007/s00217-012-1848-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-012-1848-y