
Abstract
The present study describes an event-specific quantitative Real-time PCR detection method for the transgenic Bt rice line Kemingdao 1 (KMD1). This rice line which is not approved in any country so far is likely to be approved in China in the near future. The developed primers amplify a DNA sequence spanning the integration site of the genetic construct in KMD1. DNA sequence information of this unknown site necessary for primer design was achieved using SiteFinding-PCR technique. The specificity of the detection method was shown by testing a number of different transgenic and conventional plant varieties (e.g. rice LL 601, LL 62, Bt 63). As alternative to genomic DNA, we developed double target hybrid amplicons as synthetic calibration standards in Real-time PCR. These amplicons contained both one copy of the KMD1 event-specific sequence and one copy of a sequence of the rice reference gene gos9. The limit of quantification (LOQ) of the method was tested to be 0.05%.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 2007, transgenic plants were grown in 23 countries on an estimated area of 114 million ha with soybean, maize, cotton, and canola being the most important crops [1]. Despite the efforts made in the development of transgenic rice lines, there has been no large-scale commercial cultivation so far. The acceptance of the farmers associations, exporting companies and consumers, as well as the public debates and concerns on safety aspects are still very controversial.
Since the first transformation of rice in 1988 [2] and the achievement of fertile transgenic rice plants [3], some genetically modified rice lines were developed with traits like herbicide tolerance [4, 5], resistance against bacteria [6, 7] or viruses [8, 9] or nutritional alteration [10–12], while insect resistance conferred by so called cry-genes from Bacillus thuringiensis—often referred to as “Bt” lines—is the most important trait (for excellent overview see reviews by [13, 14]. A trend in the development of future genetically modified rice lines is also the generation of stacked genes for the simultaneous expression of, e.g. disease and insect resistance [15].
In some important rice producing Asian countries like the Philippines [16] and India [17, 18], experiments with transgenic rice are still carried out in the greenhouse. Various field trials were already conducted for the evaluation of field performance and safety assessment of genetically modified rice lines in China [19–21], Pakistan [22], Iran [23], Costa Rica [24], and the Mediterranean region [25, 26]. Furthermore, several studies are carried out on the risk assessment of Bt rice concerning food safety aspects [21, 28–30].
At present, only few transgenic rice varieties are approved for commercial cultivation (LL 62, LL 06 and LL 601 in the USA [31]) and Bt rice in Iran, whereas Iran is the only country in which transgenic rice is grown at a semi-commercial level [1]. China, the world’s major rice producer, has approved no transgenic rice variety so far, although China has made great efforts in the development of transgenic rice and has conducted extensive field testings of more than 100 transgenic rice lines during the past decade [19, 32–34]. China is likely to be the first Asian country to approve genetically modified (GM) rice, which would then result in the world’s first large-scale cultivation of a genetically modified crop for direct human consumption and would have a major impact on the production of GM rice in other Asian countries [14, 35].
As the area of cultivated transgenic plants is growing continuously, the risk of contaminations with unauthorised GM material is increasing as demonstrated by the discovery of traces of the at that time unauthorised LL601 rice in EU rice imports from the USA [36] or the transgenic rice line Shanyou 63 (Bt63) rice from China [37] in the food chain. The discovery of US rice contaminated with LL rice 601 forced the European Union to take emergency measures which require that imports of specific rice products be proved by an accredited laboratory to be free of contaminations with LL rice 601 [38]. Furthermore, there is concern that seeds from field trials with GM rice were propagated and are planted illegally by Chinese farmers [39].
In practice, competent authorities in charge of food and feed control are facing the problem that for routine testing and surveillance for Genetically Modified Organisms (GMO) in Europe generally only the detection methods for approved GMO are available, and that for a rising number of non-approved (in the EU) GMOs those methods are not at hand. However, precautionary consumer protection often demands that also non-approved GMOs can be detected, e.g. by official control authorities. Therefore, we developed an event-specific Real-time PCR detection method for the transgenic Chinese Bt rice line Kemingdao 1 (KMD1) which is currently tested in extensive field trials in China [20, 40, 41]. Due to its agronomic performance and resistance to eight lepidopteran pest species like the yellow stem borer, the striped stem borer and the rice leaf folder [19, 20, 32], KMD1 has the potential to be approved in China in the near future, as lepidopteran insects frequently cause severe yield losses in rice producing countries [42, 43].
In Japan, Bt rice DNA with a construct similar to the Kemingdao line could be detected in rice vermicelli (rice noodles) products imported from China [44]. A clear identification of the corresponding GMO, however, was not possible.
Due to the lack of available information about the sequence of the genetic construct in KMD1, we used the SiteFinding-PCR method [45] for gene walking. This technique allowed the amplification of unknown DNA sequences and the characterisation of the DNA flanking the insertion site of the newly introduced gene construct. The DNA sequence data obtained were used for the development of an event-specific Real-time PCR detection method for the transgenic rice line KMD1.
Material and methods
Plant material
Seeds of the transgenic rice line Kemingdao 1 (KMD1) and the non-modified isogenic line Xiushui 11 (X11) were kindly provided by Prof. Dr. Qingyao Shu (Zhejiang-University, Hangzhou, China). KMD1 line was derived from the commercial japonica rice variety Xiushui 11 by Agrobacterium transformation [46]. The gene construct contains a synthetic cryIA(b) gene from B. thuringiensis under the control of the maize ubiquitin promoter, the regulatory elements P35S from Cauliflower Mosaic Virus (CaMV), Tnos from Agrobacterium tumefaciens and the selection marker genes nptII (neomycin phosphotransferase gene) and hpt (hygromycin phosphotransferase gene) linked in tandem [47, 48]. Molecular characterisation via Southern blot showed that KMD1 is homozygous for the transgene and has one single insertion of the gene construct [32, 40, 49].
DNA extraction
Rice grains were mechanically disrupted with sterile pestle and mortar. DNA was extracted with the Wizard Kit (Promega) according to the manufacturer’s protocol. Purity and quantity of the DNA were measured by UV spectrophotometer (Ultraspec 3000, Pharmacia Biotech) and PicoGreen® analysis (Genios Plus, Tecan).
Determination of unknown DNA sequences by SiteFinding-PCR
No sequence information was available concerning the insertion site of the genetic construct in KMD1. For sequence determination, unknown DNA fragments flanking the insert had to be amplified. For that purpose the SiteFinding-PCR was used. SiteFinding-PCR is a fast method for gene or chromosome walking [45]. The so called SiteFinder is a single-stranded molecule of 61 bp in length comprising a GCGC nucleotide sequence motive at its 3′-end followed by six degenerated bases for unspecific binding (Table 1). The SiteFinder anneals to unknown DNA regions and serves as target for the SiteFinder Primers (SFP) which together with gene-specific primers (GSP), binding to a known sequence, allow the amplification of an unknown DNA fragment.
The binding position of the used Sitefinder primers 1 and 2 (SFP1 and SFP2) at the SiteFinder molecule is shown in Fig. 1.

Sequence of the SiteFinder molecule and the binding sites of the SiteFinder Primers SFP1 and SFP2 [45]

Sequence of the hybrid amplicon gos9-KM and primers and probes for the Real-time PCR method. Reference gene gos9 target sequence (1–68 bp), adapter sequence (69–88 bp), and GMO target sequence (89–166 bp)
Since the transformation of KMD1 rice line is based on the transformation vector pKUB [47] and thus on the binary vector pBin19 [46], the Binv5 primer, which is suited for the amplification of pBin19 based T-DNA inserts (Qingyao Shu, personal communication), was used as starting point for the amplification of an unknown sequence near the presumed integration site of the gene construct.
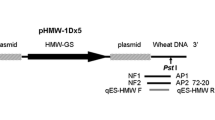
For the determination of the region spanning, the 3′-end of the inserted DNA and the host plant DNA and the design of the gene-specific primers (GSP) for the SiteFinding-PCR, in a first step a PCR was performed with the primer pair Binv5/HA-nos118-f (Table 1) binding in the pBin19 binary vector and in the Tnos sequence of the gene construct, respectively (Fig. 3).

Schematic illustration of the genetic construct in KMD1 according to [47] (a) and the transition range from rice host DNA to the transformation vector pBin19 determined in this study (b). The position of the primer/probe system KM2_for, KM1_rev and KM-p used for the event-specific detection of KMD1 is shown by arrows. Primers HA-nos 118-f and Binv5 was used to amplify and determine a sequence near the integration site for the design of gene-specific primers for the SiteFinding PCR
The PCR was run in a total volume of 25 μl with 12.5 μl HotStarTaq Mastermix (Qiagen), 0.4 μM of each Primer (Binv5 and HA-nos118-f) and 40 ng DNA with the following cycling conditions: 95 °C for 15 min, 39 cycles with 95 °C for 30 s, 60 °C for 30 s., 72 °C for 45 s, and a final elongation step at 72 °C for 3 min. After visualisation of the PCR fragments by agarose gel electrophoresis and ethidium bromide staining for 20 min, the PCR product was purified with the PCR Product Purification Kit (Qiagen) and sequenced. DNA sequencing was performed on an ABI Prism™ 310 Genetic Analyzer platform (Applied Biosystems) using the Big Dye Terminator Kit (Applied Biosystems). Sequence analysis and alignment were made using the software SeqMan (DNASTAR, Madison, USA).
Sequence data were used for the design of gene-specific primers GSP1 and GSP2 for the SiteFinding-PCR.
SiteFinding-PCR
The hybridisation of the SiteFinder molecule to the template DNA of KMD1 was performed in a total volume of 20 μl with the following reagents: 12.5 μl HotStarTaq MasterMix (Qiagen), 50 nM SiteFinder primer and 40 ng genomic KMD1 DNA. One single PCR cycle was performed as follows: 94 °C for 15 min, 92 °C for 2 min, 95 °C for 1 min, 25 °C for 1 min, ramping to 72 °C over 3 min, and 72 °C for 10 min.
After the first PCR cycle, 1.0 μl HotstarTaq Mastermix (Qiagen), 1 μl GSP1 (10 pmol/μl), 1 μl SFP1 (50 pmol/μl), and 2 μl H2O were added to the PCR mix on ice. The PCR conditions for the subsequent amplification of the region between GSP1 and SFP1 were as follows: 1 min at 94 °C, 30 cycles with 95 °C for 10 s, 58 °C for 3 min and a final elongation at 72 °C for 5 min. The PCR product was diluted 1:100 with 1 × TE buffer (pH 8.0) and subsequently used as template in a nested PCR. The nested PCR was performed in a total volume of 50 μl with 25 μl HotstarTaq Mastermix (Qiagen), 0.2 μM GSP2, 0.2 μM SFP2, and 2 μl template DNA. A nested PCR was run in parallel with the same PCR conditions, but using GSP3 instead of GSP2. The cycling conditions of the nested PCR were: 94 °C for 15 min, 30 cycles 95 °C for 10 s, 58 °C for 3 min, and a final elongation at 72 °C for 5 min. PCR products were purified using the PCR Product Purification Kit (Qiagen) according to the manufacturer’s protocol and subsequently sequenced.
Generation of genomic DNA standards and samples
For quantification of GMO ratios in DNA samples by quantitative Real-time PCR genomic standards were generated by serial dilution of KMD1 DNA with 0.2 × TE buffer. The calibration standards nominally contained 10,000, 1,000, 250, 50, and 20 copies for the transgene detection method of KMD1 (KM) and 40,000, 20,000, 10,000, 5,000, and 1,000 copies for the reference gene gos9, respectively.
DNA Samples for quantification were prepared by dilution of KMD1 DNA with DNA from non-modified (conventional) rice. The samples nominally contained 5.0, 1.0, 0.5, 0.1, and 0.05% of genetically modified genomic DNA from rice line KMD1.
Production of hybrid amplicons
In order to circumvent the problem of limited supply of genomic reference DNA of KMD1, double target hybrid amplicons containing both one copy of the KMD1 event-specific transition sequence and one copy of a sequence of the rice reference gene gos9 were generated for use as synthetic standard in Real-time PCR [52, 54].
To generate hybrid amplicons, the reference gene gos9 [51] and the target sequence of the transgene construct were first amplified in separate PCR runs using 25 μl Hotstart Mastermix (Qiagen), 0.2 μM of each primer (org1/org2_ov and KM2_f_ov/KM1_rev) and 20 ng KMD1 rice DNA in a total volume of 50 μl. PCR conditions were: 15 min heat activation at 96 °C, 39 cycles with 30 s at 94 °C, 30 s at 61.6 °C, 30 s at 72 °C, and a final elongation step at 72 °C for 3 min. The adapter primers (org2_ov and KM2_f_ov) thereby generated an elongation of 20 bp on the PCR products with complementary sequences.
After purification with the QIAquick PCR Purification Kit (Qiagen), the gos9 and KM PCR products were diluted 1:100 with 1 × TE and mixed 1:1. Two microlitres of the mixture served as template in the subsequent PCR for the generation of the hybrid amplicons using the primer combination org1 and KM1_rev. With the adapter sequence of these two primers, the two PCR products (KM and gos9) were connected to form one molecule. To reduce the amount of single PCR amplicons that have not been assembled, the amplified hybrid amplicon was purified as described above and used subsequently as template for a reamplification with the same primers and reaction conditions. The theoretically expected length of the hybrid amplicon of 166 bp (reference gene gos9, 68 bp; gene construct KM, 78 bp; adapter sequence, 20 bp) was experimentally confirmed by sequence analysis (Fig. 2).
Before their use as calibration standards, the hybrid amplicons were diluted with 0.2 × TE buffer to a concentration of 10,000, 1,000, 250, 50, 20, and 10 copies for KM and 40,000, 10,000, 1,000, 250, and 50 copies for gos9, respectively.
Real-time PCR
Based on the DNA sequence of the identified integration site of the construct into the rice genome, an event-specific Real-time PCR method for the detection of KMD1 was designed. The positions of primers and probe are shown in Fig. 4. PCR Primers and probe were designed using the software Primer Express 2.0 (Applied Biosystems). As reference method for the quantification of GMOs, the rice specific genes gos9 [51] and RPD (Rice Phospholipase D) [53] were used.

DNA sequence of the pBin19 and rice host genome transition range found in rice line KMD1. Gene-specific primers GSP1, GSP2 and GSP3 are underlined. The primers for the event-specific Real-time PCR method (KM2_for and KM1_rev) are boxed, the probe (KM-p) is written in italics. The Binv5 Primer is underlined with dots. Vector DNA sequence (red) ranges from 1–269 bp, host rice DNA sequence starts from 270 to 556 bp
Real-time PCR reactions were run on an ABI PRISM 7900 HT (Applied Biosystems) using the following programme: 2 min at 50 °C, 10 min at 95 °C, 45 cycles of 20 s at 95 °C and 1 min at 60 °C. PCR reactions were performed in a 25 μl reaction mix with 12.5 μl 2 × TaqMan Universal Mastermix (Applied Biosystems), 5 μl DNA (20 ng/μl), 300 nM of each of the primers org1 and org2, 100 nM probe orgp for the quantification of gos9 and 300 nM KM2_for, 900 nM KM1_rev and 100 nM probe KM_p for the detection of KMD1, respectively. For the amplification of the RPD gene, 200 nM of each of the primers KVM159 and KVM160 and the probe TM013 were used. All samples were measured in triplicates.
Results and discussion
Amplification of unknown DNA from rice line KMD1 and sequence analysis
Due to the lack of sequence information concerning the integration site of the genetic construct in KMD1, unknown DNA sequences had to be characterised. Using the primers HA-nos118-f and Binv5, a 704-bp fragment of the transition range from the Tnos sequence to the transformation vector was generated. Based on the sequence information of this PCR fragment, the gene-specific primers (GSP1, GSP2 and GSP3) for the subsequent SiteFinding-PCR were designed. The application of the SiteFinding-PCR facilitated the amplification of unknown DNA sequences and a step-by-step approaching towards the integration site and the rice (host) DNA.
A sequence alignment of the SiteFinding-PCR products by BLAST search (http://www.ncbi.nlm.nih.gov, BLASTN 2.2.18+) identified the integration site of the genetic construct in KMD1. This transition sequence was deposited in GenBank (EU980363.1). Figure 4 illustrates the sequence of the adjacent Oryza sativa DNA (AP008208.1) and the binary transformation vector pBin19 (AY995145.1) on which the transformation vector of KMD1 pKUB is based [47].
SiteFinding-PCR has previously been demonstrated by other authors to be a suitable method for the amplification of unknown DNA sequences [45, 58]. Due to its unspecific binding site the SiteFinder primer anneals at frequently occurring short repetitive GCGC segments. However, different from a potential amplification length of up to 4 kb in Arabidopsis [45] PCR fragments obtained in rice only reached an average length of approximately 300 bp. Due to this limitation in fragment length the SiteFinding-PCR had to be repeated several times with new gene-specific primers.
Specificity of the Real-time PCR
For the event-specific detection of KMD1, the region spanning the transition from rice DNA to the binary vector pBin19 was chosen for the development of a Real-time PCR method.
The PCR primers KM2-f and KM1-rev amplify a 78-bp fragment, thus also allowing the amplification of strongly fragmented DNA, e.g. in processed food and feed. To increase the specificity of the Real-time PCR, the probe was placed exactly at the junction location of the inserted construct. PCR analysis with DNA from different transgenic and conventional plant varieties (rice, maize, oilseed rape, and soybean) showed that the Real-time PCR is specific for the detection of KMD1 (Table 2), as no amplification signals were obtained with any other tested DNA sample. The amplifiability of the tested DNA was previously checked by PCR using plant specific primers (detection of reference genes, data not shown).
Real-time PCR analysis
All calibration standards prepared with genomic DNA (gDNA) for the detection of KMD1 (KM) and the reference genes gos9 and RPD and the standards prepared with the hybrid amplicon containing the KM and gos9 target sequence showed PCR efficiencies ranging from 90 to 97.8%. The calibration standards resulted in calibration lines with a regression coefficient R 2 of about 0.99 (Table 3; Fig. 5).

Amplification plot of the event-specific Real-time PCR method for the detection of KMD1 with a dilution series of genomic KMD1 DNA. Standards were diluted to a concentration of 10,000, 1,000, 250, 50, and 20 copies (nominal) per reaction. Calibration curves were produced by plotting the Ct-values determined for the KM-specific target against the logarithm of the copy numbers in the respective standard dilution and used for the quantification of unknown samples
Accuracy and precision
Rice DNA samples with different ratios (5, 1, 0.5, 0.1, 0.05%) of KMD1 were used in order to determine the accuracy and precision of the quantitative Real-time PCR. Accuracy was calculated as the deviation of the detected mean values from the expected values (Table 4). Precision was estimated by the relative standard deviation (rel. SD, standard deviation divided by the mean value, given in %). The calculated accuracy is in accordance with the method performance requirements of the European Network of GMO Laboratories [55], which demand that the accuracy should be within ±25% of the accepted reference value over the whole dynamic range (Table 5).
Determinations of GMO contents in DNA samples with different GMO ratios were carried out using calibration standards prepared from genomic DNA (gDNA). The rice specific genes gos9 and RPD were used as reference genes for calibration. Additionally, a hybrid amplicon containing the sequence of the event-specific detection method KM for KMD1 and the target sequence of gos9 was developed and tested for its suitability for quantification. Other studies have already shown that the rice reference genes gos9 and RPD can be used for accurate quantification of transgenic rice [51, 53].
Our experiments showed that the use of hybrid amplicons as calibration material for the quantification of KMD1 is a good alternative to genomic reference material. The advantage of hybrid amplicons is that they in general can easily be produced and handled by any GMO laboratory. Furthermore, it can help in cases where reference material (genomic DNA) is in short supply.
Limit of quantification
For the determination of the limit of quantification (LOQ), DNA of non-transgenic rice was spiked with DNA of KMD1 rice in concentrations of 5–0.05%. With a total of 35 ng DNA per reaction, the limit of quantification was at least 0.05% as this GMO ratio could still be quantified correctly. The limit of detection (LOD) was not determined but is presumably below 0.05% and depends on the quality of the extracted DNA and the food or feed matrix investigated [56]. Therefore, the Real-time PCR method complies with the Definition of Minimum Performance Requirements for Analytical Methods of GMO testing of the European Network of GMO Laboratories [55] concerning the LOQ and LOD which should currently be in the range of 0.09 and 0.045%, respectively. Taking into consideration that 35 ng of DNA was used per PCR reaction and the molecular mass of one haploid rice genome copy is 0.44 pg [57], a GMO ratio of 0.05% corresponds to approximately 40 copies of the transgene.
Conclusions
The present study describes a sensitive and reliable Real-time PCR method for the event-specific detection of the transgenic Bt rice line KMD1. Together with each of the two tested rice reference genes gos9 and RPD, the assay is suited to detect and quantify traces of KMD1 in rice DNA samples. The production of a hybrid amplicon containing both the transgenic and a reference gene target sequence for the use as calibration standard may provide a helpful alternative to genomic DNA standards. For GMO screening, however, the hybrid amplicons cannot replace reference material like genomic DNA or multi-target plasmids, as they contain one transgenic sequence only. For general screening purposes, different PCR detection systems like for P35S and Tnos [59] were successfully tested with KMD1 DNA (data not shown).
SiteFinding-PCR proved to be a successful method for the amplification of unknown DNA sequences and the detection of the integration site of the KMD1 gene construct. Further studies have to be carried out in order to receive valuable statistical data on the method performance in terms of LOQ and LOD.
References
James C (2007). Global status of commercialized Biotech/GM Crops: 2007. ISAAA Brief No. 37. ISAAA, Ithaca, NY
Zhang W, Wu R (1988) Efficient regeneration of transgenic plants from rice protoplasts and correctly regulated expression of the foreign gene in plants. Theor Appl Genet 76:835–840
Shimamoto K, Terada R, Izawa T, Fujimoto H (1989) Fertile transgenic rice plants regenerated from transformed protoplasts. Nature 338:274–276
Datta SK, Datta K, Soltanifar N, Donn G, Potrykus I (1992) Herbicide-resistant indica rice plants from IRRI breeding line IR72 after PEG-mediated transformation of protoplasts. Plant Mol Biol 20:619–629
Oard JH, Linscombe SD, Braverman MP, Jodari F, Blouin DC, Leech M, Kohli A, Vain P, Cooley JC, Christou P (1996) Development, field evaluation and agronomic performance of transgenic herbicide resistant rice. Mol Breed 2:359–368
Song WY, Wang GL, Chen LL, Kim HS, Pi LY, Holsten T, Gardner J, Wang B, Zhai WX, Zhu LH, Fauquet C, Ronald P (1995) A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 270:1804–1806
Zhai WX, Chen CY, Zhu XF, Chen XW, Zhang DH, Li XB, Zhu LH (2004) Analysis of tDNA-Xa21 loci and bacterial blight resistance effects of the transgene Xa21 in transgenic rice. Theor Appl Genet 109:534–542
Chaogang S, Jianhua W, Guoying Z, Gang S, Baozhen P, Juanli L, Dendi J, Shenxiang C, Upadhyaya NM, Waterhouse P, Zuxun G (2003) Ectopic expression of the spike protein of rice ragged stunt Oryza virus in transgenic rice plants inhibits transmission of the virus to insects. Mol Breed 11:295–301
Sivamani E, Huet H, Shen P, Ong CA, de Kochko A, Fauquet C, Beachy RN (1999) Rice plant (Oryza sativa L.) containing rice tungro spherical virus (RTSV) coat protein transgenes are resistant to virus infection. Mol Breed 5:177–185
Ye X, Al-Babili S, Klöti A, Zhang J, Lucca P, Beyer P, Potrykus I (2000) Engineering the provitamin A (ß-Carotene) biosynthetic pathway into (carotenoid-free) rice endosperm. Science 287:303–305
Paine JA, Shipton CA, Chaggar S, Howells RM, Kennedy MJ, Vernon G, Wrigth SY, Hinchliffe E, Adams JL, Silverstone AL, Drake R (2005) Improving the nutritional value of Golden Rice through increased pro-vitamin A content. Nat Biotechnol 23(4):482–487
Potrykus I, Burkhardt PK, Datta SK, Fütterer J, Ghosh Biswas GC, Klöti A, Spangenberg G, Wünn J (1995) Genetic engineering of indica rice in support of sustained production of affordable and high quality food in developing countries. Euphytica 85:441–449
Tyagi A, Mohanty A (2000) Rice transformation for crop improvement and functional genomics. Plant Sci 158:1–18
Bajaj S, Mohanty A (2005) Recent advances in rice biotechnology—towards genetically superior transgenic rice. Plant Biotechnol J 3:275–307
Datta K, Baisakh N, Maung TK, Tu J (2002) Pyramiding transgenes for multiple resistance in rice against bacterial blight, yellow stem borer and sheath blight. Theor Appl Genet 106:1–8
Ghareyazie B, Alinia F, Menguito CA, Rubia LG, de Palma JM, Liwanag EA, Cohen MB, Khush GS, Bennett J (1997) Enhanced resistance to two stem borers in an aromatic rice containing a synthetic cryIA(b) gene. Mol Breeding 3:401–414
Khanna HK, Raina SK (2002) Elite indica transgenic rice plants expressing modified Cry1Ac endotoxin of Bacillus thuringiensis show enhanced resistance to yellow stem borer (Scirpophaga incertulas). Transgenic Res 11:411–423
Ramesh S, Nagadhara D, Pasalu IC, Kumari AP, Sarma NP, Reddy VD, Rao KV (2004) Development of stem borer resistant transgenic parental lines involved in the production of hybrid rice. J Biotechnol 111:131–141
Ye GY, Shu QY, Yao HW, Cui HR, Cheng XY, Hu C, Xia YW, Gao MW, Altosaar I (2001) Field evaluation of resistance of transgenic rice containing a synthetic cry1Ab gene from Bacillus thuringiensis-Berliner to two stem borers. J Econ Entomol 94:271–276
Ye GY, Yao HW, Shu QY, Cheng X, Hu C, Xia YW, Gao MW, Altosaar I (2003) High levels of stable resistance in transgenic rice with a cry1Ab gene from Bacillus thuringiensis Berliner to rice leaffolder, Cnaphalocrocis medinalis (Guenée) under field conditions. Crop Protection 22:171–178
Wang Z, Wang Y, Cui H, Xia Y, Altosaar I, Shu Q (2002) Toxicological evaluation of transgenic rice flour with a synthetic cryIAb gene from Bacillus thuringiensis. J Sci Food Agric 82:738–744
Bashir K, Husnain T, Fatima T, Latif Z, Mehdi SA, Riazuddin S (2004) Field evaluation and risk assessment of transgenic indica basmati rice. Mol Breed 13:301–312
Mousavi A, Malboobi MA, Esmailzadeh NS (2007) Development of agricultural biotechnology and biosafety regulations used to assess the safety of genetically modified crops in Iran. J AOAC Int 90(5):1513–1516
Espinoza-Esquivel AM, Arrieta-Espinoza G (2007) A multidisciplinary approach directed towards the commercial release of transgenic herbicide-tolerant rice in Costa Rica. Transgenic Res 16:541–555
Breitler JC, Vassal JN, Catala MDM, Meynard D, Marfà V, Mele E, Royer M, Murillo I, Segundo SB, Guiderdoni E, Messeguer J (2004) Bt rice harbouring cry genes controlled by a constitutive or wound-inducible promoter, protection and transgene expression under Mediterranean field conditions. Plant Biotechnol J 2:417–430
Marfà V, Melé E, Gabarra R, Vassal JM, Guiderdoni E, Messeguer J (2002) Influence of the developmental stage of transgenic rice plants (cv. Senia) expressing the cry1B gene on the level of protection against the striped stem borer (Chilo suppressalis). Plant Cell Rep 20:1167–1172
Wu D, Shu Q, Ye Q, Zhang L, Ma C, Xia Y (2003) Comparative studies on major nutritional components and physicochemical properties of the transgenic rice with a synthetic cry1Ab gene from Bacillus thuringiensis. J Food Biochem 27:295–308
Poulsen M, Kroghsbo S, Schrøder M, Wilcks A, Jacobsen H, Miller A, Frenzel T, Danier J, Rychlik M, Shu Q, Emami K, Sudhakar D, Gatehouse A, Engel KH, Knudsen I (2007) A 90-day safety study in Wistar rats fed genetically modified rice expressing snowdrop lectin Galanthus nivalis (GNA). Food Chem Toxicol 45:350–363
Poulsen M, Schrøder M, Wilcks A, Kroghsbo S, Lindecrona RH, Miller A, Frenzel T, Danier J, Rychlik M, Shu Q, Emami K, Taylor M, Gatehouse A, Engel KH, Knudsen I (2007) Safety testing of GM-rice expressing PHA-E lectin using a new animal test design. Food Chem Toxicol 45(2007):364–377
Schrøder M, Poulsen M, Wilcks A, Kroghsbo S, Miller A, Frenzel T, Danier J, Rychlik M, Emami K, Gatehouse A, Shu Q, Engel KH, Altosaar I, Knudsen I (2007) A 90-day safety study of genetically modified rice expressing Cry1Ab protein (Bacillus thuringiensis toxin) in Wistar rats. Food Chem Toxicol 45:339–349
APHIS (Animal and Plant Health Inspection Service) (2007) http://www.aphis.usda.gov/
Shu Q, Ye G, Cui H, Cheng X, Xiang Y, Wu D, Gao M, Xia Y, Hu C, Sardana R, Altosaar I (2000) Transgenic rice plants with a synthetic cry1Ab gene from Bacillus thuringiensis were highly resistant to eight lepidopteran rice pest species. Mol Breed 6(4):433–439
Tu J, Zhang G, Datta K, Xu C, He Y, Zhang Q, Khush GS, Datta SK (2000) Field performance of transgenic elite commercial hybrid rice expressing Bacillus thuringiensis δ-endotoxin. Nat Biotechnol 18:1101–1104
Wang Y, Johnston S (2007). UNU-IAS Working Paper No. 152. Review on GM rice risk assessment in China. http://www.ias.unu.edu/resource_centre/152%20Yanqing%20Wang_1.pdf
Jia H, Jayaraman KS (2004) China ramps up efforts to commercialize GM rice. Nat Biotechnol 22(6):642
Vogel G (2006) Genetically modified crops. Tracing the transatlantic spread of GM rice. Science 313:1714
Mäde D, Degner C, Grohmann L (2006) Detection of genetically modified rice: a construct-specific Real-time PCR method based on DNA sequences from transgenic Bt rice. Eur Food Res Technol 224:271–278
European Commission (2006) Commission decision of 5 September 2006 on emergency measures regarding the non-authorised genetically modified organism “LL RICE 601” in rice products (2006/601/EC)
Zi X (2005) GM rice forges ahead in China amid concerns over illegal planting. Nat Biotechnol 23(6):637
Wang Z, Shu Q, Gongyin Y, Cui H, Wu D, Altosaar I, Xia Y (2002) Genetic analysis of resistance of Bt rice to stripe stem borer (Chilo suppressalis). Euphytica 123:379–386
Wang H, Ye Q, Wang W, Wu L, Wu W (2006) Cry1Ab protein from Bt transgenic rice does not residue in rhizosphere soil. Environ Pollut 143:449–455
Alam MF, Datta K, Abrigo E, Vasquez A, Senadhira D, Datta SK (1998) Production of transgenic deepwater indica rice plants expressing a synthetic Bacillus thuringiensis cryIA(b) gene with enhanced resistance to yellow stem borer. Plant Sci 135:25–30
High SM, Cohen MB, Shu QY, Altosaar I (2004) Achieving successful deployment of Bt rice. Trends Plant Sci 9(6):286–292
Akiyama H, Sasaki N, Sakata K, Ohmori K, Toyota A, Kikuchi Y, Watanabe T, Furui S, Kitta K, Maitani T (2007) Indicated detection of two unapproved transgenic rice lines contaminating vermicelli products. J Agric Food Chem 55:5942–5947
Tan G, Gao Y, Shi M, Zhang X, He S, Chen Z, An C (2005) SiteFinding-PCR: a simple and efficient PCR method for chromosome walking. Nucleic Acid Res 33(13):e122
Cheng X, Sardana R, Kaplan H, Altosaar I (1998) Agrobacterium-transformed rice plants expressing synthetic cryIA(b) and cryIA(c) genes are highly toxic to striped stem borer and yellow stem borer. PNAS 95:2767–2772
Wu G, Cui H, Ye G, Xia Y, Sardana R, Cheng X, Li Y, Altosaar I, Shu Q (2002) Inheritance and expression of the cry1Ab gene in Bt (Bacillus thuringiensis) transgenic rice. Theor Appl Genet 104:727–734
Babekova R, Funk T, Pecoraro S, Engel K-H, Baikova D, Busch U (2008) Duplex polymerase chain reaction (PCR) for the simultaneous detection of cryIA(b) and the maize ubiquitin promoter in the transgenic rice line KMD1. Biotechnol Biotechnol Equip 22(2):705–708
Gatehouse AMR, Emami K, Raemakers R, Shu QY (2004). Production and characterisation of transgenic rice lines. In: Knudsen I, Poulsen M, Kledal TS (eds) SAFOTEST. New methods for safety testing of transgenic food.
§ 64 LFBG. Amtliche Sammlung von Untersuchungsverfahren nach § 64 Abs. 1 LFGB. Bundesamt für Verbraucherschutz und Lebensmittelsicherheit (Herausgeber), Beuth Verlag GmbH, Berlin
Hernández M, Esteve T, Pla M (2005) Real-time polymerase chain reaction based assays for quantitative detection of barley, rice, sunflower, and wheat. J Agric Food Chem 53:7003–7009
Pardigol A, Guillet S, Pöpping B (2003) A simple procedure for quantification of genetically modified organisms using hybrid amplicon standards. Eur Food Res Technol 216:412–420
CRL (2006) Community reference laboratory for GM food feed. Event-specific method for the quantitation of rice line LLRICE62 using Real-time PCR. http://gmo-crl.jrc.it/
Moreano F, Pecoraro S, Bunge M, Busch U (2005) Development of synthetic DNA-standards for the quantitative screening of different genetically modified rapeseed lines via Real-time PCR. Proceedings of the Euro food chem XIII conference, Hamburg, Germany, 21–23 September 2005, pp 154–158
ENGL (European Network of GMO Laboratories) (2005) Definition of minimum performance requirements for analytical methods of GMO testing. http://gmo-crl.jrc.ec.europa.eu/doc/Min_Perf_Requir_Analyt_methods_131008.pdf
Charels D, Broeders S, Corbisier P, Trapmann S, Schimmel H, Linsinger T, Emons H (2007) Toward metrological traceability for DNA fragment ratios in GM quantification. 2. Systematic study of parameters influencing the quantitative determination of MON 810 corn by Real-time PCR. J Agric Food Chem 55:3258–3267
Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Rep 9:208–218
Zhao G, Zhang Z, Sun H, Li H, Dai H (2007) Isolation of Ty1-copia-like retrotransposon sequences from the apple genome by chromosome walking based on modified SiteFinding-polymerase chain reaction. Acta Biochim Biophys Sin 39(9):675–683
Lipp M, Bluth A, Eyquem F, Kruse L, Schimmel H, Van den Eede G, Anklam E (2001) Validation of a method based on polymerase chain reaction for the detection of genetically modified organisms in various processed foodstuffs. Eur Food Res Technol 212:497–504
Acknowledgments
This work was funded by the Bavarian State Ministry of the Environment, Public Health and Consumer Protection.
Author information
Authors and Affiliations
Corresponding author
Additional information
R. Babekova and T. Funk contributed equally to this paper.
Rights and permissions
About this article
Cite this article
Babekova, R., Funk, T., Pecoraro, S. et al. Development of an event-specific Real-time PCR detection method for the transgenic Bt rice line KMD1. Eur Food Res Technol 228, 707–716 (2009). https://doi.org/10.1007/s00217-008-0981-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-008-0981-0