
Abstract
The acreage for genetically modified crops (GMOs)—particularly soybean—has steadily increased since 1996, when the first crop of Roundup Ready soybean (intended for food production) was grown. The Roundup Ready soybean varieties derive from a soybean line into which a glyphosate-resistant enolpyruvylshikimate-3-phosphate-synthase (EPSPS) gene was introduced. The inserted and the flanking regions in Roundup Ready soybean have recently been characterized. It was shown that a further 250-bp fragment of the epsps gene is localized downstream of the introduced nos terminator of transcription, derived from the nopaline synthase gene from Agrobacterium tumefaciens. We examined whether this 250-bp fragment could be of functional importance. Our data demonstrate that at least 150 bp of this DNA region are transcribed in Roundup Ready soybean. Transcription of the fragment depends on whether read-through events ignore the nos terminator signal located upstream. Our data also indicate that the read-through product is further processed, resulting in four different RNA variants from which the transcribed region of the nos terminator is completely deleted. Deletion results in the generation of open reading frames which might code for (as yet unknown) EPSPS fusion proteins. The nos terminator is used as a regulatory element in several other GMOs used for food production. This implies that read through products and transcription of RNA variants might be a common feature in these GMOs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genetically modified Roundup Ready soybean (RRS) and varieties derived thereof have been approved for food and animal feed production since 1996 in the European Union. In 2003, genetically modified soybeans—predominantly RRS—accounted for more that 50% of the worldwide soybean production [1]. All RRS varieties derive from event GTS 40-3-2, which was created by ballistic methods that introduced the glyphosate-resistant CP4-enolpyruvylshikimate-3-phosphate-synthase (CP4 EPSPS) gene from Agrobacterium tumefaciens strain CP4 into the genome of the soybean [2, 3]. Transcription of CP4 EPSPS is controlled by a CaMV-promoter and the nos terminator derived from Cauliflower Mosaic Virus and the nopaline synthase gene from A. tumefaciens, respectively.
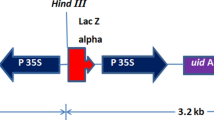
The genomic organisation and flanking regions of the insert in RRS event GTS 40-3-2 were characterized recently. It was shown that a further 250-bp fragment of the cp4 epsps gene was introduced downstream of the inserted cp4 epsps gene and nos terminator region [4] (see Fig. 1). Read-through mechanisms of terminator regions of transcription are known and essential to the life cycles of several viruses, for example [5–8]. The aim of the present study was to examine whether read-through of the nos terminator takes place in the context of the full-length cp4 epsps gene in RRS, which might result in transcription of the 250 bp EPSPS repeat. The presented data demonstrate read-through of the nos terminator and at least partial transcription of the epsps-gene fragment located downstream of the terminator region. Furthermore, our data indicate that the read-through transcript is posttranscriptionally processed, resulting in the generation of RNA variants from which both the UGA stop codon of the CP4 EPSPS open reading frame (ORF) and the transcribed nos terminator region were completely removed. In silco analysis revealed that these RNA variants might code for (as yet unknown) CP4 EPSPS fusion proteins.

Scheme for the CP4 EPSPS insert in Roundup Ready soybean and the primers used in the study. Shown is the genomic DNA (gDNA) region with the CaMV-promoter, nos terminator (filled circles), the ORF of the EPSPS gene (in gray) and the two DNA repeat regions 1 and 2 located within the EPSPS gene (repeat 1) and downstream of the nos terminator (repeat 2). Relative locations of the binding sites of the primers used for PCR analysis are indicated by arrows. Transcripts initiating from the CaMV-promoter proceeding up to or beyond the nos terminator of transcription are indicated below (dashed lines). Only read-through transcripts beyond the nos terminator in RRS should result in amplification products with the primer pairs A (PP-A), B (PP-B), and C (PP-C) via RT-PCR analysis. The length of the expected amplification products with the corresponding primer pairs, using gDNA from RRS as the template for PCR is indicated
Material and methods
Isolation of genomic DNA and RNA from soybean leaves
The RRS (line GTS 40-3-2) and the corresponding isogenic wildtype soybean was provided by Monsanto Agrar Deutschland GmbH [9]. 9–15 days after germination, leaves of the planted soybeans were harvested for analysis. For DNA and total RNA isolation, 500 mg of tissue from leaves were shock frozen in liquid nitrogen, homogenized with a pestle and mortar in TriPure Isolation Reagent (Roche), and prepared according to the protocol supplied. Isolated RNA was treated with 50 U RNase free DNase I (Roche) for 30 min at 37 °C to remove any genomic DNA which might be present within the samples. Thereafter, the RNA was purified by phenol/chloroform extraction, washed twice in 75% ethanol, precipitated, and stored at −80 °C until further use.
Reverse transcription of prepared RNA samples
Complementary DNA (cDNA) was transcribed from 2.5 μg of total RNA from RRS, and the corresponding wildtype soybean initiated with random nonamer primer and Superscript II reverse transcriptase (100 U, Invitrogen) for 2 h at 39 °C following the manufacturer’s recommendations. As a control, identical RNA-probes were treated accordingly but without addition of the reverse transcriptase. Thereafter, cDNAs were purified with AutoSeq G-50 columns (Amersham Biosciences) and used in PCR analysis.
PCR analysis and description of primers used
All PCRs contained 1 U Taq DNA Polymerase (Roche) in 10 mM Tris–HCl (pH 8.3), 1.5 mM MgCl2, 50 mM KCl, 200 μM each dNTP, and 0.2 μM of forward and reverse primers in 50 μl volume. PCR conditions were 35 cycles of 20 s at 94 °C, 20 s at 55 °C, and 1.5 min at 72 °C, with 4 min at 94 °C prior to the reaction and 7 min of final extension at 72 °C in a Mastercycler gradient cycler (Eppendorf). PCR products were resolved in a 1% agarose gel and stained with ethidium bromide. Either ~50 ng of genomic DNA or 10% of the volume of the reverse transcriptase reaction was used as template.
Nucleotide sequences of the primers used for PCR analysis:
-
GM01f (forward): 5′-TGCCGAAGCAACCAAACATGATCCT-3′
-
GM02r (reverse): 5′-TGATGGATCTGATAGAATTGACGTT-3′
-
P32 (forward): 5′-GAATCCTGTTGCCGGTCTTG-3′
-
P33 (reverse): 5′-TTATCCTAGTTTGCGCGCTA-3′
-
P150 (reverse): 5′-AGGTGTCGCCTTCCTTACG-3′
-
P-149 (forward): 5′-TCGTGTCGGAAAACCCTG-3′
-
P123r (reverse): 5′-TGGCGCCCATGGCCTGCATG-3′
The primers GM01f and GM02r correspond to sequences in the soybean lectine gene and they amplified a DNA fragment of 120 bp. All other primers used are specific to the insert in RRS. The relative locations of the binding sites for these primers and the lengths of the expected PCR products are shown in Fig. 1.
Isolation and cloning of PCR products
DNA fragments were cut out of the agarose gel and isolated with the QIAquick Gel Extraction Kit (Qiagen) according to the supplied protocol. The isolated fragments were concentrated in a speed vac (Concentrator 5301, Eppendorf) and ligated into the vector pGEM (pGEM Teasy Vector System, Promega) according to the protocol supplied.
Sequencing of the isolated cDNA clones
Plasmid DNAs with the inserted cDNAs were isolated with the QIAprep Spin Miniprep Kit (Qiagen) as recommended in the manual supplied. The nucleotide sequences of the cDNAs were determined at the MWG Biotech Company for both strands with the sequencing primers T7 5′-TAA TAC GAC TCA CTA TAG GG-3′ and Sp-6 5′-CGA TTT AGG TGA CAC TAT AG-3′. Sequence alignments were performed with the MacVector software (Oxford Molecular Group).
Results and discussion
To analyze potential read-through transcripts which represent the 250 bp EPSPS repeat located downstream of the nos terminator, RNA was prepared from the genetically modified RRS and the corresponding wildtype soybean. To ensure that genomic DNA was completely removed from the isolated RNA before reverse transcription, a PCR was performed with the cDNAs and primers specific to the soybean lectine gene (GM01f/GM02r). As expected, a DNA fragment with a length of 120 bp was amplified using genomic DNA isolated from leaves of RRS and the corresponding wildtype (data not shown). Identical amplification products of 120 bp were detected in the samples with the cDNAs as template. No PCR products were detectable in control samples prepared as described above without addition of reverse transcriptase (data not shown). This result indicates that genomic DNA was completely removed from the RNA samples used for cDNA synthesis.
We then analyzed whether expression of the introduced CP4 EPSPS gene proceeds beyond the nos termination signal of transcription. Read-through of the nos terminator may result in production of an over-length RNA, reflecting the introduced EPSPS repeat region inserted downstream of the nos terminator. Therefore, PCR analysis was performed with cDNAs prepared from RRS or the corresponding wildtype, and the primer pairs PP-A (P32 and P33) or PP-B (P32 and P150). As expected from the published sequence information about the insert introduced into RRS (accession no.: AY125353) amplification products of about 177 bp and 362 bp were detected with the primer pairs P32f/P33r and P32f/P150r, respectively, using genomic DNA prepared from RRS (see Fig. 2, lanes 4, 5). Identical PCR products were detected with the cDNA from RRS as template (see Fig. 2, lanes 8, 9). In controls with cDNAs from wildtype soybean, or with samples not treated with reverse transcriptase, no amplification products were detectable (Fig. 2, lanes 6, 7, 10–13). These results indicate that (in RRS at least) the 5′ region of the EPSPS repeat region is transcribed. To confirm the observation that read-through of transcription occurs beyond the nos terminator, in the region located downstream thereof, RT-PCRs were performed with a further primer pair. Analysis of genomic DNA from RRS as template with the primer pair PP-C (P-149 and P123) resulted in amplification of a single PCR product of about 540 bp, consistent with the published sequence information (Fig. 3, lane 3). An identical PCR product was also detected using cDNA from RRS as template (Fig. 3, lane 5). These results demonstrate that the DNA-region inserted downstream of the nos terminator is transcribed in RRS. Additional amplification products of lower molecular weights were also detected. To characterize these products, the amplified fragments were isolated and ligated into the vector pGEM. Thereafter, the sequences of the cloned cDNA-fragments were determined and are depicted in Fig. 4. The determined sequences of the cloned cDNA fragments were aligned against the corresponding genomic sequence of RRS (accession no.: AY125353). The data indicate that the read-through transcript was processed in four different RNA-variants, from which both the UGA stop codon of the CP4 EPSPS ORF and the complete transcribed nos terminator region were removed (Fig. 4). From these RNA variants, regions of 377 bases (cDNA clone #3), 401 bases (cDNA clone #4), 368 bases (cDNA clone #5), and 413 bases (cDNA clone #6) were deleted. The variants from which the nos terminator regions were detected were found via RT-PCR with the primer pair C only (Fig. 3, lane 5). PCR analysis of the cDNAs from RRS with the primer pairs A or B revealed the unprocessed read-through but no amplification products with lower molecular weight (Fig. 2, lanes 8, 9), since the binding site of the forward primer used is located within the region of the deleted nos terminator.

Detection of read-through transcripts beyond the nos terminator of transcription in RRS. Genomic DNA (gDNA) and RNA were isolated from RRS and the corresponding wildtype (WT). The isolated RNAs were treated with RNase free DNase I to remove gDNA, and reverse transcribed into cDNAs initiated with random nonamer primers. Identical RNA-probes used as controls were also treated, but without addition of reverse transcriptase (RT). PCR was performed with the primer pairs A (PP-A) or B (PP-B) and the templates (templ.) indicated. Using gDNA prepared from RRS, amplification products of 177 bp for PP-A or 362 bp for PP-B were expected (see Fig. 1). A 100-bp molecular weight standard was used as marker (M)

Detection of RT-PCR products derived from read-through transcripts beyond the nos terminator in RRS. The experiment was performed as described in the legend to Fig. 2, except that the primer pair C (PP-C) was used. PCR with gDNA and PP-C is expected to result in an amplification product of 541 bp. A 50-bp molecular weight standard was used as marker (M)

Alignment of the DNA sequences of the isolated cDNA clones with the corresponding genomic region from RRS. The sequences of the cDNA clones (cDNA #3, #4, #5, #6) were aligned to the sequence for the insert in RRS (accession no.: AY125353). For the genomic RRS insert (gDNA) the region from nucleotide position (ntd.-pos.) 1359 to 1901 is shown. The TGA stop codon of translation of the EPSPS ORF (at ntd.-pos. 1480, bold) and the region of the introduced nos terminator (ntd-pos. 1531-1831, italicized) are indicated. The binding sites of the primers P-140 and P-123, used for PCR amplification, are underlined. The nucleotides deleted from the described RNA variants are shown as dashes
The RNA variants are probably generated posttranscriptionally, by unknown mechanisms. Canonical splicing mechanisms do not appear to mediate the generation of these RNA variants, since the sequences at the cleavage sites of the described RNA variants do not correspond to the consensus sequence said to be involved in splicing in plants [10, 11]. However, we cannot rule out that these RNA variants contain cryptic or uncommon sites which might be recognized by factors of the splicing machinery. The cis regulatory regions that initiate and mediate splicing are located within the removed region of spliced transcripts [10]. If this is also true for the mechanisms mediating posttranscriptional processing of the described variants, it seems reasonable to assume that the transcribed nos terminator region might be responsible for processing the RNA. Since the nos terminator was and still is commonly used as regulatory region in the production of genetically modified crops (http://agbios.com/dbase.php), read-through products and RNA variants could also be expressed in these plants. It would be interesting to test this assumption with characterized GMOs that also contain the nos terminator as regulatory region.
Since the stop codon of the CP4 EPSPS ORF was deleted for the variants, ORFs with modified N-termini are expected. Therefore, we analyzed the processed read-through transcripts for ORFs which might serve as a template for translation based on our and other published sequence data. Although different regions were deleted from the described cDNA clones, the ORFs of all variants proceeded beyond the junction in one frame into the EPSPS repeat region. In this way, the OFRs of the variants encode for CP4 EPSPS fusion proteins, each with a redundancy of at least 56 amino acids at the N-terminus identical to amino acid region 60–115 of the CP4 EPSPS protein (see Fig. 5). Moreover, the ORFs of all variants contain a further region of 24 amino acids derived from the region adjacent to the insert derived from the host genome. For this N-terminal region, no sequence homology was found to proteins published in the NCBI database. Thus, the revealed ORFs potentially code for CP4 EPSPS fusion proteins with identical N-termini, that differ in the region of junction only. As deduced from our data, the RNA variants described in this study may code for as yet unknown CP4 EPSPS fusion proteins.

Potential open reading frames of the RNA variants. Analysis for ORF was performed based on published (accession no.: AY125353) and our DNA-sequence data. The complete amino acid sequence of the CP4 EPSPS protein is shown. The sequences of the potential CP4 EPSPS fusion proteins start at amino acid position 401. The redundant regions at the N-terminus of the CP4 EPSPS fusion proteins and the corresponding region within the CP4 EPSPS protein are underlined. The N-terminal amino acid region encoded by the plant genome adjacent to the introduced insert in RRS is indicated in (bold)
Taken together, our data demonstrate that the nos terminator signal of transcription introduced into the genome in RRS is (at least in part) ignored, resulting in the production of an over-length transcript. Furthermore, this transcript was found to be processed posttranscriptionally, resulting in the production of different RNA variants. These variants might code for as yet unknown CP4 EPSPS fusion proteins with a common N-terminus of 24 amino acids with no homology to proteins of the NCBI database. Since the nos terminator was introduced as regulatory region in several other GMOs, read-through products and RNA variants might be transcribed in these transgenic crops as well.
References
James C (2003) Preview: global status of commercialized transgenic crops: 2003, ISAAA briefs no. 30. ISAAA, Ithaca, NY
Nair RS, Fuchs RL, Schuette SA (2002) Toxicol Pathol 30:117–125
Padgette SR, Kolacz KH, Delannay X, Re DB, LaVallee BJ, Tinius CN, Rhodes WK, Otero YI, Barry GF, Eichholtz DA, Peschke VM, Nida DL, Taylor NB, Kishore GM (1995) Crop Sci 35:1451–1461
Windels P, Taverniers I, Depicker A, Van Bockstaele E, De Loose M (2001) Eur Food Res Technol 213:107–112
Nassal M, Schaller H (1993) Trends Microbiol 1:221–228
Wang WK, Chen MY, Chuang CY, Jeang KT, Huang LM (2000) J Microbiol Immunol Infect 33:131–140
Roebuck KA, Saifuddin M (1999) Gene Expr 8:67–84
Zhao J, Hyman L, Moore C (1999) Microbiol Mol Biol Rev 63:405–445
Federal Institute for Risk Assessment (1998) § 35 LMBG L-23.01.22. Federal Institute for Risk Assessment, Berlin
Brown JWS (1996) Plant J 10:771–780
White O, Soderlund C, Shanmugan P, Fields C (1992) Plant Mol Biol 19:1057–1064
Acknowledgements
We would like to thank Thomas Kammertöns and Monika Schwarz for support and valuable discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rang, A., Linke, B. & Jansen, B. Detection of RNA variants transcribed from the transgene in Roundup Ready soybean. Eur Food Res Technol 220, 438–443 (2005). https://doi.org/10.1007/s00217-004-1064-5
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-004-1064-5