
Abstract
A robust capillary electrophoresis (CE) method for the determination of the six main organic acids in wines is described. The CE method is based on the use of 2,6- pyridinedicarboxylic acid (PDC) as background electrolyte (BGE) for the indirect UV detection of tartaric, malic, citric, succinic, acetic and lactic acids. Ethylenediaminetetracarboxylic acid (EDTA) was added to the BGE to eliminate interferences from trace metals on the determination of citric acid. Problems related to linearity reported in the literature using the same BGE were shown to be the result of the sample pH being lower than that of the buffer. Although this could be overcome simply by adjusting the pH of the sample, electrokinetic injection was found to be a better alternative, improving at the same time the sensitivity of the determination. Linearity over the range 100–5,000 mg/l (ppm) for all acids was achieved, requiring only 80× dilution of standards and samples prior to injection. Within day repeatabilitys (n=7) of migration times (RSD%<0.15) and peak areas (RSD%<5) as determined for a wine sample were found to be excellent. The method was applied to 20 South African wine samples, and the results are compared to data obtained by ion-exclusion liquid chromatography. Succinic acid was consistently overestimated using the latter technique, while for the other acids good agreement between the methods was noted.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Organic acids play a major role in the microbiological and physicochemical stability as well as in the sensory properties of wines [1]. Knowledge of the specific organic acid content of grapes, musts and wines is important to the winemaker because the data provide valuable information on the optimum harvest period, the progress of fermentation and the overall quality of the wine. For these reasons a fast and reliable analytical method to determine organic acids is of utmost importance for the wine industry.
Various modes of liquid chromatography (LC) have been used for the analysis of organic acids. Reversed phase LC (RPLC) with UV detection [2, 3, 4, 5, 6, 7, 8] suffers from poor sensitivity and selectivity, and in most cases derivatisation is required thereby increasing the total analysis time. Better separation is obtained using ion-exclusion chromatography (IEC) [9, 10, 11, 12, 13], in which the analytes are separated by a combination of ion-exclusion and partitioning processes. UV and/or refractive index (RI) are the detection methods of choice for IEC. However, expensive columns and harsh operating conditions do detract from the overall usefulness of the method. Moreover, in the case of complex samples such as red wines, sample clean-up is required for reliable quantitation.
Capillary electrophoresis (CE) has increasingly been shown to be an attractive alternative to LC for the analysis of natural products [14, 15]. Advantages over LC include increased speed and efficiency of separation and low column costs and sample/solvent consumption. The analysis of organic acids by capillary zone electrophoresis (CZE) with indirect UV detection has received a lot of attention [16, 17, 18, 19, 20, 21, 22, 23]. The choice of background electrolyte (BGE) is of crucial importance, and several alternatives have been proposed [15, 20]. Soga and Ross introduced 2,6-pyridinedicarboxylic acid (PDC) as BGE for the determination of organic and inorganic anions, amino acids and carbohydrates in a variety of natural samples [21, 22]. The method has the advantage of being fast, sensitive, reliable and quantitative. Moreover, the effect of metals in the system that might interfere with the analysis of some organic acids is masked. Kandl and Kupina applied this method to the analysis of wine and grape juice [23]. The method allowed accurate determination of organic acids with only sample dilution prior to injection. However, severe restrictions regarding the linearity for acetic and succinic acid were reported.
In this contribution the phenomenon responsible for the non-linearity is elucidated and an improved robust CE method employing a PDC-based BGE for the determination of organic acids in wines is presented.
Materials and methods
Materials
l-Malic acid, tartaric acid, ethelenediaminetetracarboxylic acid (EDTA), cetyltrimethylammonium bromide (CTAB) and sodium azide were purchased from Sigma-Aldrich (Atlasville, South Africa). Acetic acid and lactic acid were from Riedel-de Haën (Midrand, South Africa), succinic acid (disodium salt), formic acid and 2,6- pyridinedicarboxylic acid (PDC) from Acros (Geel, Belgium) and citric acid from Merck (Darmstadt, Germany). 20 mM sulphuric acid (Merck) used as mobile phase for IEC was filtered through 0.45-µm HV filters before use (Millipore Corporation, Bedford, MA). Buffers for CE were filtered through disposable syringe filters (Millex HN 0.45-µm Nylon, Millipore). The SPE cartridges (Strata SDB-L styrene-divinylbenzene, 3 ml, 500 mg phase) were from Phenomenex (Torrance, Calif., USA). The wine samples were purchased from local stores. If not analysed immediately, the samples were transferred under nitrogen to completely filled amber bottles. 20-µM sodium azide was added to the mobile phase for IEC and BGE for CE as well as to the standard solutions.
Instrumentation
CE and data analyses were performed on an HP3D CE capillary electrophoresis system equipped with diode array detection (Agilent Technologies, Waldbronn, Germany). Bare fused silica capillaries (Composite Metal Services Ltd., Worcester, UK) with an internal diameter of 75 µm and total lengths of 80 or 110 cm were used. In the final conditions a 7.5 mM PDC buffer containing 0.5 mM CTAB and 0.5 mM EDTA was prepared by dissolving the appropriate amounts and adjusting the pH to 5.60 with 1 M NaOH. Injection was performed either hydrodynamically by applying a pressure of 50 mbar for 3 s or electrokinetically by applying −10 kV for 2 s. In both cases a buffer plug was injected (50 mbar, 2 s) after the sample. Between runs the capillary was flushed for 4 min with buffer, followed by a voltage conditioning step at −15 kV for 1 min. The separation voltage was ramped from 0 to −22 kV in 0.5 min. Indirect UV detection was performed at 350 nm with a reference wavelength of 210 nm. The capillary temperature was set at 15 °C.
IEC analyses were carried out on an Alliance 2690 separations module equipped with a 996 photodiode array detector (Waters, Milford, MA, USA). Data analysis was done using Millenium32 Chromatography Manager software (Waters). The column (Aminex HPX-87H ion exclusion column, 300 mmx7.8 mm) was equipped with a guard column of the same phase (Bio-Rad, Nazareth, Belgium). The mobile phase was 20 mM H2SO4. All experiments were performed at a flow rate of 0.6 ml/min. The injection volume was 10 µl and the column temperature was kept constant at 50 °C. Detection was performed at 210 nm.
Preparation of standard solutions and samples for CE
Formic acid was used as internal standard for both CE and LC experiments. Calibration samples of 100, 500, 1,000, 2,500 and 5,000 mg/l (ppm) of all six acids were prepared in deionised water. The calibration samples were diluted 80× with a 25.32-ppm formic acid solution to produce samples containing 25 ppm of the internal standard. Wine samples were diluted in the same manner.
Preparation of standard solutions and samples for IEC
The same calibration levels were used in LC, without dilution. Each sample contained 1,000 mg/l (ppm) formic acid as internal standard. The wine samples were fractionated by SPE. The styrene-divinylbenzene cartridges were conditioned with 3 ml each of ethyl acetate, methanol and water (pH 2.5). The pH of each wine sample was adjusted to 2.5 with 6 M HCl prior to spiking with 5,000 ppm formic acid. A 1-ml sample was loaded onto the cartridges. The organic acids were rinsed of with 4×1 ml of 20-mM sulphuric acid, and a 50-µl sample of the combined (ca. 5 ml) eluent was injected. Recoveries determined using an artificial wine sample were used for quantitative calculations.
Results and discussion
A BGE consisting of PDC at pH 5.60 and containing 0.5 mM CTAB provides sufficient resolution and sensitivity for the analysis of the six major wine acids [23]. When determining the optimum buffer concentration, the following aspects had to be considered: the concentration of the buffer (and visualising ion) should be kept at a minimum in order to reduce baseline noise, while at the same time being sufficient high to ensure effective separation, sensitivity and buffering capacity. Kandl et al. [23] considered 5 mM PDC to be the minimum concentration needed for reproducible migration times. In this study, formic acid was used as internal standard in both the CE and LC analyses. It was found that a PDC concentration of 7.5 mM was needed to provide baseline separation between formic and tartaric acids. For this BGE the best signal-to-noise ratios for the acids were obtained by using a signal wavelength of 350 nm with a reference wavelength of 210 nm. Capillaries of 75-µm i.d. with lengths of 80 or 110 cm and a working temperature of 15 °C were chosen in order to achieve acceptable sensitivity while limiting the generated current (and concurrent Joule heating). Although the six wine acids could be separated under these conditions, two problems were encountered: the citric acid peak displayed severe tailing, increasing its limit of detection and complicating quantitation, and, as reported by Kandl et al. [23] detector response for acetic and succinic acids was only linear up to 750 and 1,500 ppm, respectively.
It has been reported that the presence of metal cations in the separation system does affect the migration times and peak areas of certain acids. For some BGEs, acids with high stability constants may even be lost at low concentrations [21, 24]. In the case of wine acids this will predominantly affect the analysis of citric acid because of the higher stability constant of metal complexes formed with this acid. However, Soga and Ross reported that the complexation ability of PDC is sufficient to mask the effect of trace metals present in the capillary by preferential complexation [21]. Moreover, Kandl et al. reported no such problems when they applied this method to wine analysis [23]. During initial experiments we observed badly tailing peak shapes for citric acid. Increasing the PDC concentration up to 20 mM did not improve this. In order to evaluate whether this was the result of metals present in the system, EDTA was added to the BGE. Because of the high stability constant of EDTA–metal complexes, this should remove any interferences from metals. Indeed, peak height for citric acid increased with the EDTA concentration from 0.05 to 0.5 mM, giving a perfect peak shape at 0.5 mM. Consequently 0.5 mM was chosen as the optimum concentration. Contrary to previous reports, the complexing ability of PDC is insufficient (up to the 20-mM level) to eliminate interference from metal cations. The effect of addition of EDTA to the BGE on the peak shape of citric acid is illustrated in Fig. 1.

Effect of EDTA on the peak shape of citric acid. a 7.5 mM PDC, 0.5 mM CTAB, pH 5.6. b 7.5 mM PDC, 0.5 mM CTAB, 0.5 mM EDTA, pH 5.6. Sample: 1,000 ppm organic acid standards, diluted 40×. Capillary: 75 μm i.d., 80 cm L tot. Injection: 50 mbar for 3 s. Other conditions as in text. Peaks: 1=formic acid (I.S.), 2=tartaric acid, 3=malic acid, 4=citric acid, 5=succinic acid
The lower linear range for acetic and succinic acid compared to the other acids was ascribed by Kandl and Kupina [23] to be the result of a “saturation effect to effectively displace the visualising ion”. We observed similar problems during initial experiments employing pressure injection. It was noted that the peaks for these two acids became severely distorted to the point of being split at the higher calibration levels. In addition, when the calibration graphs for these acids were constructed individually (without the addition of the other five acids), the responses were linear over the whole range. Initially we thought this was a result of mismatch in conductivities between the sample plug and the BGE, with the former being significantly higher at the higher calibration levels. This could cause a form of de-stacking, where those solutes in the buffer accelerate away from the sample plug (this process is the opposite to that which occurs during field-amplified sample stacking), leading to the observed split peaks. However, this would not explain why the effect is only significant for acetic and succinic acids. In other words, the difference in mobility in the sample and buffer regions is greater for these two acids than for the others. This can be explained by looking at the pH of the sample and the pKa values of the acids (Table 1). Since the pH of the sample is lower than that of the buffer, most acids will have higher charge-to-mass ratios in the buffer region. Acetic and succinic acid have the highest pKa1 values of all the acids. Accordingly, only a small percentage of these acids is charged in the sample region, whereas both possess at least a −1 charge in the buffer. This translates into large difference in electrophoretic mobilities (μe) of these acids between the two regions, and can explain why split peaks are observed for these acids (those solutes in the buffer region accelerate away from the sample zone). The rest of the acids are all ionised to a greater extent in the sample zone, and thus the difference in mobilities between the two phases is not as pronounced. The effect will become more important as the pH of the sample drops with increasing acid content, which explains why the problem does not occur at lower calibration levels or when these acids are calibrated individually.
Figure 2 demonstrates the effect of sample pH on the peak shape of succinic (peak 5) and acetic acid (peak 6). A 5,000-ppm calibration sample was diluted 40× and injected in the pressure mode with the pH unadjusted at 3.39 (A) and with the pH adjusted to 5.6 (B). The disappearance of split peaks for both acids is evident. To our knowledge, this is the first time that the effect of sample pH on the de-stacking of solutes has been reported. The phenomenon also suggests the opportunity for stacking solutes by increasing the pH of the sample, in cases where the solutes are only partially charged in the BGE.

Effect of sample pH on the peak shape of acetic and succinic acids. Sample: 5,000 ppm organic acid standards, 40× diluted. a pH unadjusted at 3.39 b pH adjusted to 5.6. BGE: 7.5 mM PDC, 0.5 mM CTAB, 0.5 mM EDTA, pH 5.6. Capillary: 75 μm i.d., 110 cm L tot. Other conditions as in Fig. 1. Peaks: 1=formic acid (I.S.), 2=tartaric acid, 3=malic acid, 4=citric acid, 5=succinic acid. 6=acetic acid, 7=lactic acid, *=EDTA system peak
As an alternative to adjusting the sample pH, we investigated the use of electrokinetic injection. During this mode of injection, the solutes are loaded onto the capillary by a combination of their electrophoretic mobilities and the electro-osmotic flow (EOF). Under the operating conditions of the method, i.e. reversed EOF, the electrophoretic mobilities of the organic acids are in the same direction as the EOF. It was found that the amount of organic acids loaded onto the capillary when employing −10 kV for 2 s, is significantly higher than for pressure injection. Also, the responses for all the acids were linear up to 2,500 ppm. When using an 80× instead of a 40× dilution, linear responses for all six acids over the whole calibration range were obtained, while simultaneously improving the limits of detection (LOD). Peak shapes for acetic and succinic acid were normal over the whole range. The problems encountered with pressure injection are most likely overcome by a combination of two factors: the further dilution increases the sample pH, while the lower applied voltage during electrokinetic injection reduces the difference in the velocity of acetic and succinic acid between the sample and buffer zones (since v=μe E). Finally, the repeatability of the optimised method was investigated by injecting seven dilutions of the same wine sample. The figures of merit are summarised in Table 2. No influence of the wine matrix was observed on the migration times and peak shapes at the dilutions applied.
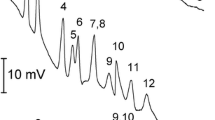
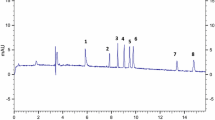
The developed CE method was used to analyse 20 South African red and white wines. The results were compared to those obtained for the same wines using ion-exclusion chromatography. Examples of an electropherogram and chromatogram for a red wine are depicted in Figs. 3 and 4, respectively. From these figures it is clear that the baseline separation of all acids obtained by CE cannot be matched by the LC method. The sensitivity of the latter is better but sample preparation is required.

Optimised CZE separation of South African wine (Red Blend 2002b, diluted 80×). Injection: −10 kV, 2 s. Other conditions as in Fig. 2

Ion-exclusion LC analysis of Red Blend 2002b, after SPE clean-up. Conditions as specified in text. Peak numbering as in Fig. 2
Quantitative results using both methods are summarised in Table 3. With a few exceptions the quantitative data are in good agreement. For tartaric acid, excellent agreement between both methods is achieved. No over-estimation of tartaric acid by CE was observed for these wines, as was reported by Kandl and Kupina [23] because of co-elution of fumaric acid. For malic and citric acids, good agreement at higher concentrations is noted. For wines containing these acids in small amounts, the values measured by IEC are generally higher, mainly due to difficult integration of the small peaks in the chromatogram. Succinic acid was constantly overestimated by the LC method and this by a factor of 3 to 10. From the chromatogram and the UV spectrum of the peak it was clear that this is the result of another compound co-eluting with succinic acid. Acetic and lactic acid values were consistently slightly higher by the CE method. In conclusion, the CE method was found to be more reliable, with the added advantage of not requiring a sample preparation step.
Conclusions
A reliable CE method for the determination of the six major organic acids in wines has been established. The method is based on the use of PDC as BGE. EDTA was added to improve the peak shape and quantitation of citric acid. Limitations regarding linearity were shown to be the result of a lower sample pH than that of the BGE. These problems were avoided while simultaneously increasing the LOD, by employing electrokinetic instead of pressure injection. For CE analysis only 80× dilution with an internal standard solution is required. The method delivers sufficient sensitivity and repeatability for wine analysis, and is considered, because of absence of co-elution, to be more reliable than ion-exclusion chromatography.
References
Ribereau-Gayon P, Gloris Y, Maujean A, Dubourdieu D (2000) Handbook of enology, vol 2. The chemistry of wine, stabilization and treatments. Wiley, New York, pp 3–39
Badoud R, Pratz G (1986) J Chromatogr 360:119–136
Caccamo F, Carfagnini G, di Corcia A, Samperi R (1986) J Chromatogr 362:47–53
Tusseau D, Benoit C (1987) J Chromatogr 395:323–333
Marcé RM, Calull M, Olucha JC, Borrull F, Rius FX (1991) J Chromatogr 542:277-293
García Romero E, Sánchez Muñoz G, Martín Alvarez PJ, Ibáñez MD (1993) J Chromatogr 360:111–117
Marcé RM, Calull M, Borrull F, Rius FX (1990) Am J Enol Vitic 41:289–294
Marcé RM, Calull M, Machobas RM, Borrull F, Rius FX (1990) Chromatographia 29:54–58
Frayne RF (1986) Am J Enol Vitic 37:281–287
Dewaele C, Sandra P, Vanlaer L (1991) Analusis 19:351–354
Calull M, Marcé RM, Borrull F (1992) J Chromatogr 590:215–222
Falqué López E, Fernández Gómez E (1996) J Chromatogr Sci 34:254–257
Klein H, Leubolt R (1993) J Chromatogr 640:259–270
Sádecká J, Polonský J (2000) J Chromatogr A 880:243–279
Klampfl CW, Buchberger, Haddad PR (2000) J Chromatogr A 881:357–364
Lalljie SPD, Vindevogel J, Sandra P (1993) J Chromatogr A 652:563–569
Arellano M, Couderc F, Puig Ph (1997) Am J Enol Vitic 48:408–412
Dabek-Zlotorzynska E, Piechowski M, McGrath M, Lai EPC (2001) J Chromatogr A 910:331–345
Wang M, Qu F, Shan X-Q, Lin J-M (2003) J Chromatogr A 989:285–292
Poppe H, Xu X (1998) Indirect detection in capillary electrophoresis. In Khaledi MG (Ed) High performance capillary electrophoresis, theory, techniques and applications. Chemical analysis series, vol 146. Wiley, New York, pp 375–403
Soga T, Ross GA (1997) J Chromatogr A 767:223–230
Soga T, Ross GA (1999) J Chromatogr A 837:231–239
Kandl T, Kupina S (1999) Am J Enol Vitic 50:155–161
Horie H, Yamauchi Y, Kohata K (1998) J Chromatogr A 817:139–144
Acknowledgements
A. de Villiers thanks the National Research Foundation of South Africa for a study grant. We thank the KWV (Paarl, South Africa) for financial support and Dr. C. Dewaele for donation of the IEC column.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Villiers, A., Lynen, F., Crouch, A. et al. A robust capillary electrophoresis method for the determination of organic acids in wines. Eur Food Res Technol 217, 535–540 (2003). https://doi.org/10.1007/s00217-003-0819-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-003-0819-8